RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 31/01/2014
PANTOPRAZOLE MYLAN 20 mg, comprimé gastro-résistant
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé gastro-résistant contient 22,55 mg de pantoprazole sodique sesquihydraté, équivalent à 20 mg de pantoprazole.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé gastro-résistant.
Comprimés jaunes, ovales, biconvexes, gravés PS2 à l'encre noire sur une face.
4.1. Indications thérapeutiques
Chez l'adulte et l'adolescent âgé de 12 ans et plus
Reflux gastro-sophagien symptomatique.
Pour le traitement à long terme et la prévention des récidives d'sophagite par reflux gastro-sophagien.
Chez l'adulte
Prévention d'ulcères gastro-duodénaux induits par les anti-inflammatoires non stéroïdiens non sélectifs chez des patients à risque ayant besoin d'un traitement continu par AINS (voir rubrique 4.4).
4.2. Posologie et mode d'administration
Les comprimés ne doivent pas être croqués ou écrasés. Ils doivent être avalés entiers avec un peu d'eau avant un repas.
Posologie recommandée
Chez l'adulte et l'adolescent âgé de 12 ans et plus
Reflux gastro-sophagien symptomatique
La dose orale recommandée est d'un comprimé gastro-résistante de PANTOPRAZOLE MYLAN 20 mg par jour. Une amélioration des symptômes est généralement obtenue en 2 à 4 semaines. Si cette durée n'est pas suffisante, l'amélioration des symptômes sera obtenue en général par un traitement de 4 semaines supplémentaires. Une fois les symptômes disparus, la récidive des symptômes peut être prévenue par la prise à la demande de PANTOPRAZOLE MYLAN 20 mg une fois par jour, en fonction des besoins. Si le contrôle des symptômes par le traitement à la demande n'est pas satisfaisant, la reprise d'une thérapie continue peut être envisagée.
Traitement à long terme et prévention des récidives des sophagites par reflux gastro-sophagien
Pour le traitement à long terme, une dose d'entretien d'un comprimé gastro-résistant de PANTOPRAZOLE MYLAN 20 mg par jour est recommandée, avec une augmentation à 40 mg de pantoprazole par jour en cas de récidive.
PANTOPRAZOLE MYLAN 40 mg peut être utilisé dans ce cas. Après cicatrisation, la dose peut à nouveau être ramenée à 20 mg de pantoprazole.
Chez l'adulte
Prévention d'ulcères gastro-duodénaux induits par les anti-inflammatoires non stéroïdiens (AINS) non sélectifs non sélectifs chez les patients à risque ayant besoin d'un traitement continu par AINS.
La dose orale recommandée est d'un comprimé gastro-résistant de PANTOPRAZOLE MYLAN 20 mg par jour.
Populations particulières
Chez l'enfant âgé de moins de 12 ans
L'utilisation de PANTOPRAZOLE MYLAN 20 mg n'est pas recommandée chez l'enfant de moins de 12 ans en raison de l'insuffisance des données de sécurité et d'efficacité dans cette tranche d'âge.
Insuffisance hépatique
La dose journalière de 20 mg de pantoprazole ne doit pas être dépassée chez le sujet atteint d'insuffisance hépatique sévère (voir rubrique 4.4).
Insuffisance rénale
Aucune adaptation de la dose n'est nécessaire chez le sujet atteint d'insuffisance rénale.
Sujet âgé
Aucune adaptation de la dose n'est nécessaire chez le sujet âgé.
Hypersensibilité à la substance active, aux benzimidazoles substitués, ou à l'un des excipients.
4.4. Mises en garde spéciales et précautions d'emploi
Chez les patients atteints d'insuffisance hépatique sévère, les enzymes hépatiques doivent être surveillées régulièrement pendant le traitement par pantoprazole, notamment en cas d'utilisation au long cours. En cas d'augmentation des enzymes hépatiques, le traitement doit être arrêté (voir rubrique 4.2).
Administration concomitante d'AINS
L'utilisation de PANTOPRAZOLE MYLAN 20 mg pour la prévention d'ulcères gastro-duodénaux induits par les anti-inflammatoires non stéroïdiens (AINS) non sélectifs doit être limitée à des patients ayant besoin d'un traitement continu par AINS et présentant un risque accru de développer des complications gastro-intestinales.
Le risque accru doit être évalué selon les facteurs de risques individuels tels que l'âge avancé (> 65 ans), les antécédents d'ulcère gastrique ou duodénal ou d'hémorragie digestive haute.
En cas de symptômes alarmants
En présence de tout symptôme alarmant (par exemple perte de poids involontaire importante, vomissements récurrents, dysphagie, hématémèse, anémie ou méléna) et lorsqu'un ulcère gastrique est suspecté ou présent, une affection maligne doit être écartée, car le traitement par pantoprazole peut atténuer les symptômes et retarder le diagnostic.
D'autres examens doivent être envisagés si les symptômes persistent malgré un traitement approprié.
Administration concomitante d'atazanavir
L'administration concomitante d'atazanavir est un inhibiteur de la pompe à protons est déconseillée (voir rubrique 4.5). Si l'association d'atazanavir et d'un inhibiteur de la pompe à protons est jugée indispensable, une surveillance clinique régulière (par exemple une surveillance de la charge virale) est conseillée, associée à une augmentation de la posologie de l'atazanavir à 400 mg par 100 mg de ritonavir. La dose quotidienne maximale de pantoprazole recommandée est de 20 mg.
Influence sur l'absorption de la vitamine B12
Le pantoprazole, comme tout antisécrétoire gastrique, peut diminuer l'absorption de la vitamine B12 (cyanocobalamine) en raison d'hypo- ou d'achlorhydrie. Ceci doit être pris en compte chez les patients disposant de réserves réduites ou présentant des facteurs de risque de diminution de l'absorption de la vitamine B12 lors de traitement au long cours ou si des symptômes cliniques sont observés.
Traitement à long terme
Dans le cadre du traitement au long cours, notamment lorsque sa durée excède 1 an, les patients devront faire l'objet d'une surveillance clinique régulière.
Infections bactériennes gastro-intestinales
Le pantoprazole, come tous les inhibiteurs de la pompe à protons (IPP), est susceptible d'augmenter la quantité de bactéries normalement présentes dans le tractus gastro-intestinal haut. Le traitement par PANTOPRAZOLE MYLAN peut conduire à une légère augmentation du risque d'infections gastro-intestinales provoquées par des bactéries comme Salmonella et Campylobacter.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Effets du pantoprazole sur l'absorption d'autres médicaments
En raison d'une inhibition importante et durable de la sécrétion gastrique, le pantoprazole peut réduire l'absorption de médicaments dont la biodisponibilité est pH-dépendante, comme par exemple certains antifongiques azolés, tels que le kétoconazole, l'itraconazole, le posaconazole et d'autres médicaments tels que l'erlotinib.
Traitement antirétroviral (atazanavir)
L'administration concomitante d'atazanavir et d'autres médicaments du VIH, dont l'absorption est pH-dépendante, avec les inhibiteurs de la pompe à protons peut entraîner une réduction substantielle de leur biodisponibilité et peut avoir un impact sur leur efficacité. Par conséquent, l'administration concomitante d'inhibiteurs de pompe à protons avec l'atazanavir est déconseillée (voir rubrique 4.4).
Anticoagulants coumariniques (phenprocoumone ou warfarine)
Bien qu'aucune interaction n'ait été observée lors de l'administration simultanée d'inhibiteurs de la pompe à protons et de phenprocoumone ou de warfarine au cours des études de pharmacocinétique clinique, quelques cas isolés de modification de l'International Normalised Ratio (INR) ont été rapportés, lors de leur administration simultanée, après commercialisation. En conséquence, chez les patients traités avec des anticoagulants coumariniques (par exemple phenprocoumone ou warfarine), le suivi du taux de prothrombine/INR est recommandé au début et à l'arrêt du traitement, ou en cas d'administration intermittente de pantoprazole.
Autres études d'interactions cinétiques
Le pantoprazole est largement métabolisé au niveau du foie, par le système enzymatique du cytochrome P450.
La principale voie métabolique est la déméthylation par le CYP2C19 et les autres voies métaboliques comprennent l'oxydation par le CYP3A4.
Aucune interaction cliniquement significative n'a été observée au cours d'études spécifiques portant notamment sur la carbamazépine, le diazépam, le glibenclamide, la nifédipine, et un contraceptif oral composé de lévonorgestrel et d'éthinylestradiol.
Les résultats d'une série d'études d'interactions cinétiques ont démontré que le pantoprazole n'influait pas sur les substances actives métabolisées par le CYP1A2 (comme la caféine ou la théophylline), le CYP2C9 (comme le piroxicam, le diclofénac ou le naproxène), le CYP2D6 (comme le métoprolol), ou le CYP2E1 (comme l'éthanol). Le pantoprazole n'interfère pas avec l'absorption de digoxine liée à la glycoprotéine P.
Il n'existe pas d'interactions avec les antiacides administrés de manière concomitante.
Des études d'interactions ont été menées sur l'administration concomitante de pantoprazole et de différents antibiotiques (clarithromycine, métronidazole, amoxicilline). Aucune interaction cliniquement significative n'a été démontrée.
Il y a très peu de données concernant l'utilisation du pantoprazole chez la femme enceinte. Au cours des études de reproduction chez l'animal, des signes de ftotoxicité ont été observés (voir rubrique 5.3). Le risque potentiel chez l'homme n'est pas reconnu. PANTOPRAZOLE MYLAN ne doit être utilisé au cours de la grossesse qu'en cas de réelle nécessité.
Des études chez l'animal ont montré que le pantoprazole passait dans le lait maternel. Le passage dans le lait maternel chez l'être humain a été rapporté. En conséquence, la décision de poursuivre/arrêter l'allaitement ou celle de poursuivre/arrêter le traitement par PANTOPRAZOLE MYLAN doit être prise en tenant compte du bénéfice de l'allaitement pour l'enfant et du bénéfice du traitement par PANTOPRAZOLE MYLAN chez les femmes.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables tels que sensations vertigineuses et troubles visuels peuvent survenir (voir rubrique 4.8). Les patients présentant ce type d'effet indésirable ne doivent pas conduire de véhicule ni utiliser des machines.
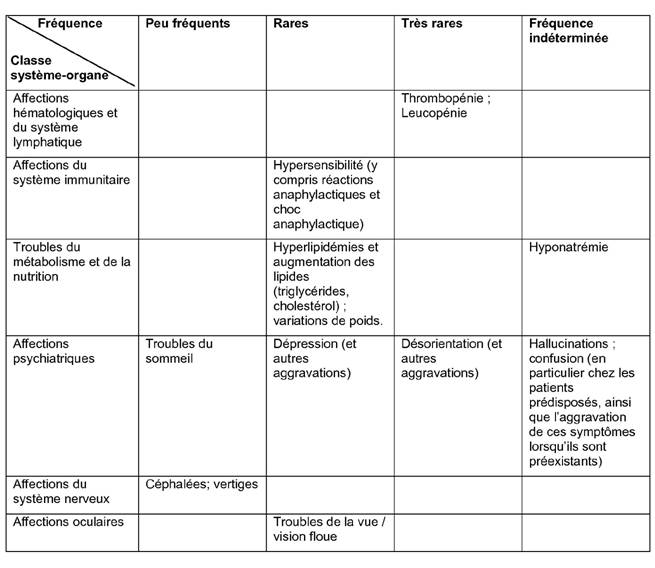
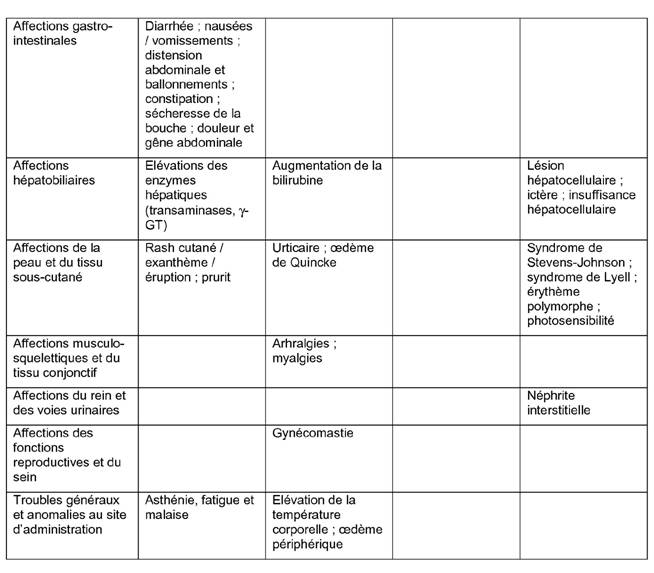
Environ 5 % des patients sont susceptibles de présenter des effets indésirables (EI). Les EI les plus souvent signalés sont une diarrhée et des céphalées, survenant tous deux chez environ 1 % des patients.
Les effets indésirables signalés avec le pantoprazole sont classés dans le tableau ci-dessous selon l'ordre de fréquence suivant:
Très fréquents (≥1/10); fréquents (≥ 1/100 à <1/10); peu fréquents (≥ 1/1 000 à < 1/100); rares (≥ 1/10 000 à <1/1 000); très rares (< 1/10 000); fréquence indéterminée (ne peut être estimée d'après les données disponibles).
Pour tous les effets indésirables notifiés après commercialisation, il n'est pas possible d'imputer cet ordre de fréquence, par conséquent ils sont listés comme survenant à une fréquence « indéterminée ».
Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Tableau 1. Effets indésirables du pantoprazole rapportés au cours des études cliniques et notifiés après commercialisation.
Les symptômes de surdosage chez l'homme ne sont pas connus.
Des doses atteignant 240 mg administrées par voie injectable en 2 minutes ont bien été tolérées. Comme le pantoprazole est largement lié aux protéines, il n'est pas aisément dialysable.
En cas de surdosage avec des signes cliniques d'intoxication, aucune recommandation thérapeutique spécifique ne peut être donnée, à l'exception d'un traitement symptomatique et de soutien.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique: INHIBITEURS DE LA POMPE A PROTONS, Code ATC: A02BC02.
Mécanisme d'action
Le pantoprazole est un benzimidazole substitué qui inhibe la sécrétion gastrique d'acide chlorhydrique de l'estomac, par un blocage spécifique des pompes à protons des cellules pariétales.
Le pantoprazole est transformé en sa forme active dans l'environnement acide des cellules pariétales où il inhibe l'enzyme H+/K+ ATPase, c'est-à-dire au niveau de la phase terminale de sécrétion d'acide chlorhydrique dans l'estomac. L'inhibition est dose-dépendante et affecte à la fois la sécrétion acide basale et la sécrétion acide stimulée. Chez la plupart des patients, la disparition des symptômes est obtenue en 2 semaines.
Comme les autres inhibiteurs de la pompe à protons, et les inhibiteurs des récepteurs H2, le traitement par pantoprazole entraîne une réduction de l'acidité de l'estomac et donc une augmentation de la gastrine proportionnelle à la diminution de l'acidité. Cette augmentation de la gastrine est réversible. Puisque le pantoprazole se lie à l'enzyme responsable de la phase terminale de la production acide, il peut inhiber la sécrétion d'acide chlorhydrique, quelle que soit la nature du stimulus (acétylcholine, histamine, gastrine). L'effet est le même, que le produit soit administré oralement ou par voie intraveineuse.
Les valeurs de gastrinémie à jeun augmentent sous pantoprazole. Dans la plupart des cas, lors des traitements de courte durée, elles ne dépassent pas les limites supérieures de la normale. Ces valeurs doublent le plus souvent lors des traitements au long cours. Toutefois, une augmentation excessive n'est rapportée que dans des cas isolés.
En conséquence, une augmentation légère à modérée du nombre des cellules endocrines de l'estomac (cellules ECL), n'est observée que dans une minorité de cas, pendant un traitement au long cours (de l'hyperplasie simple à adénomatoïde).
Cependant, d'après les études réalisées jusqu'à présent (voir rubrique 5.3), la formation de précurseurs carcinoïdes (hyperplasie atypique) ou de carcinoïdes gastriques, tels que décrits chez l'animal n'a pas été observée chez l'être humain.
Au vu des résultats des études menées chez l'animal, il n'est pas possible d'exclure totalement une influence sur les paramètres endocriniens de la thyroïde, lors de traitement au long cours de plus d'un an par pantoprazole.
5.2. Propriétés pharmacocinétiques
Absorption
Le pantoprazole est rapidement absorbé et la concentration plasmatique maximale est atteinte même après administration d'une dose orale unique de 20 mg.
En moyenne, les concentrations plasmatiques maximales, soit 1 - 1,5 µg/ml; sont atteintes en 2 heures à 2,5 heures environ après administration; ces valeurs restent ensuite constantes après administrations multiples. Les caractéristiques pharmacocinétiques ne varient pas après administration unique ou administration répétée. Dans l'intervalle de dose de 10 à 80 mg, la cinétique plasmatique du pantoprazole est linéaire après administration orale et intraveineuse.
La biodisponibilité absolue observée après la prise des comprimés est d'environ 77 %. L'administration concomitante de nourriture n'a pas d'influence sur l'ASC, sur la concentration sérique maximale ni sur la biodisponibilité. Seule la variabilité sur le temps de latence sera augmentée par une absorption concomitante de nourriture.
Distribution
La liaison aux protéines sériques de pantoprazole est d'environ 98 %. Le volume de distribution est d'environ 0,15 l/kg.
Elimination
Le principe actif est presque exclusivement métabolisé par le foie.
La principale voie métabolique est la déméthylation par le CYP2C19 sous forme de métabolites conjugués par sulfatation. Une autre voie métabolique inclut l'oxydation par le CYP3A4.
La demi-vie terminale est d'environ 1 h et la clairance est d'environ 0,1 l/h/kg. Il existe quelques cas de sujets présentant une élimination ralentie. Du fait de la liaison spécifique du pantoprazole aux pompes à protons des cellules pariétales, la demi-vie d'élimination n'est pas corrélée à la durée d'action plus longue (inhibition de la sécrétion acide).
L'élimination rénale représente la voie majeure d'excrétion (environ 80 %) pour les métabolités du pantoprazole, le reste étant éliminé dans les selles. Le métabolite principal à la fois dans le sang et les urines est le déméthylpantoprazole, sous forme sulfoconjuguée. La demi-vie du métabolite principal (environ 1,5 h) n'est pas beaucoup plus longue que celle du pantoprazole.
Caractéristiques dans des populations particulières
Environ 3 % de la population Européenne présente un déficit de fonctionnement de l'enzyme CYP2C19 et sont appelés « métaboliseurs lents ». Chez ces individus, le métabolisme du pantoprazole est principalement catalysé par le CYP3A4. Après administration d'une dose unique de 40 mg de pantoprazole, l'aire sous la courbe était en moyenne environ 6 fois supérieure chez les « métaboliseurs lents » comparativement aux sujets ayant une enzyme CYP2C19 fonctionnelle (« métaboliseurs rapides »). La concentration plasmatique maximale augmentait d'environ 60 %. Ces résultats n'ont aucune incidence sur la posologie du pantoprazole.
Aucune diminution de la dose n'est nécessaire lors de l'administration du pantoprazole chez les insuffisants rénaux (y compris les patients dialysés). Comme chez le sujet sain, la demi-vie du pantoprazole est courte. Seules de très faibles quantités de pantoprazole sont dialysées. Bien que le principal métabolite ait une demi-vie légèrement allongée (2-3 h), l'excrétion reste rapide et aucune accumulation n'est donc observée.
Malgré l'allongement de la demi-vie jusqu'à 3 à 6 h et l'augmentation de l'ASC d'un facteur 3 à 5 chez les patients cirrhotiques (classes A et B de la classification de Child), la concentration sérique maximale n'est que légèrement augmentée (x 1,3) comparativement au sujet sain.
La légère augmentation de l'ASC et de la Cmax observée chez le sujet âgé comparativement au sujet jeune n'a aucune incidence clinique.
Chez l'enfant
Après administration orale d'une dose unique de 20 ou 40 mg de pantoprazole à des enfants âgés de 5 à 16 ans, les valeurs de l'ASC et de la Cmax se sont révélées similaires à celles observées chez l'adulte. Après administration orale d'une dose unique de 0,8 ou 1,6 mg/kg de pantoprazole à des enfants âgés de 2 à 16 ans, il n'a pas été observé de corrélation significative entre la clairance du pantoprazole et l'âge ou le poids. L'ASC et le volume de distribution étaient conformes aux données observées chez l'adulte.
5.3. Données de sécurité préclinique
Les données précliniques ne mettent en évidence aucun risque particulier chez l'homme, au vu des essais pharmacologiques de sécurité, de toxicité par administrations réitérées et de génotoxicité.
Au cours des études de carcinogénicité sur 2 ans chez le rat, sont apparues des néoplasies neuro-endocriniennes. De plus, des papillomes des cellules squameuses sont apparus dans l'estomac antérieur du rat. Le mécanisme entraînant la formation de carcinoïdes gastriques par les benzimidazoles substitués a été étudié de façon approfondie et l'on peut conclure qu'il s'agit d'une réaction secondaire à l'élévation massive de la gastrinémie chez le rat au cours des études à long terme portant sur de fortes doses.
Durant les études sur 2 ans chez le rongeur, une augmentation du nombre des tumeurs hépatiques a été observée chez le rat et chez la souris femelle, et a été considérée comme due à un métabolisme hépatique important.
Une légère augmentation des transformations néoplasiques de la thyroïde a été notée dans le groupe de rats recevant la plus forte dose (200 mg/kg). L'apparition de ces néoplasies est associée aux modifications induites par le pantoprazole dans la dégradation de la thyroxine au niveau hépatique chez le rat. La dose thérapeutique chez l'homme étant faible, aucun effet sur la glande thyroïde n'est attendu.
Au cours des études de reproduction chez l'animal, des signes discrets de ftotoxicité ont été observés à des doses supérieures à 5 mg/kg.
Les études n'ont révélé aucun signe d'altération de la fertilité ou d'effet tératogène.
Le passage transplacentaire a été étudié chez le rat et semble augmenter au cours de la gestation. En conséquence, la concentration du pantoprazole chez le ftus est brièvement augmentée avant la naissance.
Noyau: carbonate de sodium anhydre (E500), mannitol (E421), crospovidone, povidone (K90), stéarate de calcium.
Composition de l'enrobage gastro-résistant:
EUDRAGIT L 30 D-55 [Laurylsulfate de sodium, polysorbate 80, copolymère d'acide méthacrylique et d'acrylate d'éthyle], triéthylcitrate.
Composition du pelliculage: OPADRY Yellow (OY-52945) [hypromellose, macrogol 400, dioxyde de titane (E171), oxyde de fer jaune (E172)].
Composition de l'encre: Opacode Black IH (S-1-27794) [Shellac (E904), oxyde de fer noir (E172), hydroxyde d'ammonium].
Sans objet.
Flacon (HDPE) : A conserver 100 jours après la première ouverture.
6.4. Précautions particulières de conservation
Films thermosoudés:
Pas de conditions particulières de conservation.
Flacons (HDPE):
Conserver les flacons soigneusement fermés à l'abri de l'humidité.
6.5. Nature et contenu de l'emballage extérieur
14, 28, 30, 50, 56, 60, 90, 98, 100, 250 comprimés en flacon (HDPE) blanc avec bouchon à vis contenant une capsule de dessicant (LDPE).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Pas d'exigences particulières.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
MYLAN SAS
117, ALLEE DES PARCS
69800 SAINT PRIEST
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 416 573-2 ou 34009 416 573 2 0: 14 comprimés sous films thermosoudés avec dessicant (Aluminium/Aluminium).
· 416 574-9 ou 34009 416 574 9 8: 28 comprimés sous films thermosoudés avec dessicant (Aluminium/Aluminium).
· 273 697-5 ou 34009 273 697 5 9 : 30 comprimés sous films thermosoudés avec dessicant (Aluminium/Aluminium).
· 416 575-5 ou 34009 416 575 5 9: 56 comprimés sous films thermosoudés avec dessicant (Aluminium/Aluminium).
· 579 697-2 ou 34009 579 697 2 1: 96 comprimés sous films thermosoudés avec dessicant (Aluminium/Aluminium).
· 584 897-6 ou 34009 584 897 6 1 : 98 comprimés sous films thermosoudés avec dessicant (Aluminium/Aluminium).
· 273 680-5 ou 34009 273 680 5 9 : 14 comprimés en flacon (HDPE).
· 273 681-1 ou 34009 273 681 1 0 : 28 comprimés en flacon (HDPE)
· 416 576-1 ou 34009 416 576 1 0: 30 comprimés en flacon (HDPE).
· 416 577-8 ou 34009 416 577 8 8 : 50 comprimés en flacon (HDPE).
· 273 682-8 ou 34009 273 682 8 8 : 56 comprimés en flacon (HDPE)
· 416 578-4 ou 34009 416 578 4 9 : 60 comprimés en flacon (HDPE).
· 579 698-9 ou 34009 579 698 9 9 : 90 comprimés en flacon (HDPE).
· 584 895-3 ou 34009 584 895 3 2 : 98 comprimés en flacon (HDPE).
· 579 699-5 ou 34009 579 699 5 0: 100 comprimés en flacon (HDPE).
· 579 700-3 ou 34009 579 700 3 1: 250 comprimés en flacon (HDPE).
· 278 003-1 ou 34009 278 003 1 3 : 7 comprimés sous films thermosoudés sans dessicant (Aluminium/Aluminium).
· 278 004-8 ou 34009 278 004 8 1 : 14 comprimés sous films thermosoudés sans dessicant (Aluminium/Aluminium).
· 278 005-4 ou 34009 278 005 4 2 : 28 comprimés sous films thermosoudés sans dessicant (Aluminium/Aluminium).
· 278 006-0 ou 34009 278 006 0 3 : 30 comprimés sous films thermosoudés sans dessicant (Aluminium/Aluminium).
· 278 007-7 ou 34009 278 007 7 1 : 56 comprimés sous films thermosoudés sans dessicant (Aluminium/Aluminium).
· 586 361-6 ou 34009 586 361 6 5 : 96 comprimés sous films thermosoudés sans dessicant (Aluminium/Aluminium).
· 586 362-2 ou 34009 586 362 2 6 : 98 comprimés sous films thermosoudés sans dessicant (Aluminium/Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste II.