|
Figure 1
|
Figure 2
|
Figure 3
|
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 22/04/2016
DELTIUS 10 000 UI/ml, solution buvable en gouttes
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
1 goutte contient 200 UI de cholécalciférol (vitamine D).
Pour la liste complète des excipients, voir rubrique 6.1.
Solution huileuse, limpide, incolore à jaune verdâtre, exempte de particule solide visible et/ou de précipité.
4.1. Indications thérapeutiques
Prévention et traitement de la carence en vitamine D.
Comme adjuvant à un traitement spécifique destiné aux patients atteints dostéoporose présentant une carence en vitamine D ou un risque de carence en vitamine D.
4.2. Posologie et mode d'administration
1 goutte contient 200 UI de vitamine D.
Population pédiatrique
· Prévention de la carence chez les nourrissons de 0 à 1 an : 400 UI/jour (2 gouttes).
· Prévention de la carence chez les enfants et adolescents de 1 à 18 ans : 600 UI/jour (3 gouttes).
· Des doses allant jusquà 1 000 UI/jour (5 gouttes) peuvent être nécessaires pour prévenir la carence chez certains enfants.
· Traitement de la carence chez les nourrissons, enfants et adolescents de 0 à 18 ans : 2 000 UI/jour (10 gouttes) pendant 6 semaines, suivi dun traitement dentretien de 400 à 1 000 UI/jour (2 à 5 gouttes).
Femmes enceintes et allaitantes
· Prévention de la carence : 400 UI/jour (2 gouttes).
· Des doses allant jusquà 2 000 UI/jour (10 gouttes) peuvent être nécessaires pour prévenir la carence chez certaines femmes (voir ci-dessous).
· Des doses encore plus élevées peuvent être nécessaires au cours de lallaitement si la femme choisit de ne pas donner de supplément de vitamine D à son enfant.
Adultes
· Prévention de la carence en vitamine D : 600 à 800 UI/jour (3 à 4 gouttes). Des doses plus élevées peuvent être requises dans certaines situations (voir ci-dessous).
· Comme adjuvant à un traitement spécifique utilisé dans lostéoporose : 800 UI/jour (4 gouttes).
Certaines populations présentent un risque élevé de carence en vitamine D et peuvent nécessiter des doses plus élevée et une surveillance du taux de 25(OH)D sérique :
· individus institutionnalisés ou patients hospitalisés ;
· individus à peau foncée ;
· individus chez qui lexposition effective au soleil est limitée par lutilisation de vêtements de protection et par lutilisation constante décrans solaires ;
· individus obèses ;
· patients évalués pour une ostéoporose ;
· utilisation de certains médicaments concomitants (par ex. anticonvulsivants et glucocorticoïdes) ;
· femmes enceintes ;
· patients présentant des syndromes de malabsorption, notamment maladie inflammatoire de lintestin et maladie cliaque ;
· individus récemment traités pour une carence en vitamine D et nécessitant un traitement d'entretien.
Les doses maximales tolérables recommandées de vitamine D sont les suivantes :
· adultes, y compris femmes enceintes et allaitantes : 4 000 UI/jour ;
· enfants et adolescents de 11 à 17 ans : 4 000 UI/jour ;
· enfants de 1 à 10 ans : 2 000 UI/jour ;
· nourrissons de 0 à 1 an : 1 000 UI/jour.
Populations spéciales
· Patients insuffisants rénaux : aucun ajustement spécifique nest requis.
· Autres pathologies : chez les patients obèses, les patients atteints de syndromes de malabsorption et les patients recevant un traitement affectant le métabolisme de la vitamine D, des doses plus élevées sont requises pour le traitement et la prévention de la carence en vitamine D (2 à 3 fois supérieures).
Mode dadministration
DELTIUS doit être pris de préférence avec un repas (voir rubrique 5.2 Propriétés pharmacocinétiques, « Absorption »).
Le flacon doit être secoué avant utilisation.
DELTIUS a un goût dhuile dolive. DELTIUS peut être pris pur ou, pour faciliter sa prise, mélangé avec une cuillérée ou une petite quantité dun aliment froid ou tiède immédiatement avant utilisation. Le patient doit sassurer de prendre la dose complète.
Chez les enfants, DELTIUS peut être mélangé avec une petite quantité daliments pour enfants, de yaourt, de lait, de fromage ou dautres produits laitiers. Les parents doivent être informés du fait qu'ils ne doivent pas mettre DELTIUS dans un biberon de lait ou dans un récipient contenant des aliments mous. En effet, si lenfant ne consomme pas la portion dans son intégralité, il ne recevra pas la dose complète. Les parents doivent sassurer que leur enfant prend la dose complète. Chez les enfants qui ne sont pas allaités, la dose prescrite doit être administrée avec un repas conséquent.
Voir également rubrique 6.6, Précautions particulières délimination et manipulation.
Instructions d'utilisation
Chaque boîte contient 1 flacon et un embout compte-gouttes. Le flacon est fermé par un opercule en plastique à lépreuve des enfants. Le compte-gouttes est protégé par un cylindre en plastique. Suivre les instructions dutilisation ci-dessous :
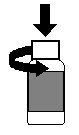
a. Pour ouvrir le flacon, appuyer sur lopercule tout en le tournant (voir Figure 1).
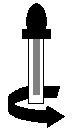
b. Avant dutiliser l'embout compte-gouttes pour la première fois, retirer le cylindre en plastique en le dévissant pour le détacher de la tige de lembout compte-gouttes (voir Figure 2).
c. Insérer lembout compte-gouttes dans le flacon pour en retirer une quantité de solution. Faire tomber le nombre de gouttes prescrites dans une cuillère.
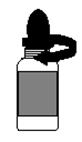
d. Pour fermer le flacon, visser soigneusement lembout compte-gouttes jusquau bout (il n'est pas nécessaire d'appuyer sur lopercule) (voir Figure 3).
e. Remettre le flacon dans son emballage dorigine.
|
Figure 1
|
Figure 2
|
Figure 3
|
Hypersensibilité au cholécalciférol ou à lun des excipients mentionnés à la rubrique 6.1.
Hypercalcémie, hypercalciurie.
Hypervitaminose D.
Calculs rénaux (lithiase rénale, néphrocalcinose) chez les patients atteints dhypercalcémie chronique au moment du traitement.
4.4. Mises en garde spéciales et précautions d'emploi
Il convient dêtre prudent chez les patients traités pour une maladie cardiovasculaire (voir rubrique 4.5 Interactions avec dautres médicaments et autres formes dinteractions, glucosides cardiotoniques y compris digitaliques).
DELTIUS doit être prescrit avec prudence chez les patients atteints de sarcoïdose, en raison dune augmentation possible du métabolisme de la vitamine D sous sa forme active. Chez ces patients, les taux de calcium urinaire et sérique doivent être surveillés.
Il convient de tenir compte de lutilisation éventuelle de traitements contenant déjà de la vitamine D, daliments enrichis en vitamine D ou de lait enrichi en vitamine D pour éviter que la dose totale de vitamine D soit trop élevée.
Si la relation de causalité entre la supplémentation en vitamine D et les calculs rénaux na pas été clairement établie, le risque est plausible, en particulier dans le contexte dune supplémentation concomitante en calcium. La nécessité dune supplémentation simultanée en calcium doit être envisagée au cas par cas. Les suppléments de calcium doivent être prescrits sous surveillance médicale étroite.
Ladministration orale de vitamine D à forte dose (500 000 UI en bolus annuel unique) augmente le risque de fractures chez le sujet âgé, en particulier au cours des 3 premiers mois suivant ladministration.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
En cas de traitement par des diurétiques thiazidiques, connus pour diminuer lélimination urinaire du calcium, une surveillance des concentrations de calcium sérique est recommandée.
Lutilisation concomitante de glucocorticoïdes peut diminuer leffet de la vitamine D.
En cas de traitement par des médicaments contenant des digitaliques et autres glucosides cardiotoniques, ladministration de vitamine D peut augmenter le risque de toxicité digitalique (arythmie). Une surveillance médicale stricte est requise, avec, si nécessaire, une surveillance des concentrations de calcium sérique et une surveillance électrocardiographique.
Un traitement simultané par une résine échangeuse dions comme la cholestyramine, le colestipol, lorlistat ou par des laxatifs comme l'huile de paraffine, peut réduire l'absorption gastro-intestinale de vitamine D.
Lagent cytotoxique actinomycine et les antifongiques imidazolés interfèrent avec lactivité de la vitamine D en inhibant la conversion de la 25-hydroxyvitamine D en 1,25-dihydroxyvitamine D par lenzyme rénale 25-hydroxyvitamine D-1-hydroxylase.
Il nexiste pas de données ou il existe des données limitées sur lutilisation du cholécalciférol chez la femme enceinte. Les études effectuées sur l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3 Données de sécurité préclinique). La dose quotidienne recommandée chez la femme enceinte est de 400 UI. Toutefois, chez les femmes considérées comme présentant une carence en vitamine D, une dose plus élevée peut être requise (jusqu'à 2 000 UI/jour, soit 10 gouttes). Pendant leur grossesse, les femmes doivent suivre les conseils de leur médecin. En effet, leurs besoins peuvent varier en fonction de la sévérité de leur pathologie et de leur réponse au traitement. La vitamine D et ses métabolites sont excrétés dans le lait maternel.
Si nécessaire, la vitamine D peut être prescrite pendant lallaitement. Cette supplémentation ne remplace pas ladministration de vitamine D au nouveau-né.
Aucun surdosage induit par la mère allaitante na été observé chez le nouveau-né. Cependant, lors de la prescription dun supplément de vitamine D à un nouveau-né allaité, le médecin doit tenir compte de la dose éventuelle de vitamine D prise par la mère.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables sont indiqués ci-dessous par classe de systèmes d'organes et par fréquence.
Les fréquences sont définies comme suit : peu fréquent (>1/1 000, <1/100) ou rare (>1/10 000, <1/1 000).
Troubles du métabolisme et de la nutrition
Peu fréquent : hypercalcémie et hypercalciurie.
Affections de la peau et du tissu sous-cutané
Rare : prurit, éruption cutanée et urticaire.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.ansm.sante.fr.
Un surdosage chronique peut provoquer une calcification vasculaire et des organes imputable à lhypercalcémie.
Traitement du surdosage
Interrompre ladministration de DELTIUS et instaurer une réhydratation.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : vitamine D et analogues, cholécalciférol, code ATC : A11CC05
Sous sa forme biologiquement active, la vitamine D stimule labsorption intestinale du calcium, lincorporation du calcium dans lostéoïde et la libération de calcium du tissu osseux. Dans lintestin grêle, elle facilite le captage rapide et différé du calcium. Le transport actif et passif du phosphate est également stimulé. Dans le rein, elle inhibe lexcrétion du calcium et du phosphate en favorisant la résorption tubulaire. La forme biologiquement active de la vitamine D inhibe directement la production de lhormone parathyroïdienne (PTH) dans les glandes parathyroïdes. La sécrétion de PTH est également inhibée par le captage accru du calcium dans lintestin grêle sous linfluence de la vitamine D sous sa forme biologiquement active.
5.2. Propriétés pharmacocinétiques
La pharmacocinétique de la vitamine D est bien connue.
Absorption
La vitamine D est bien absorbée dans le tube digestif en présence de bile. Ladministration au cours du principal repas de la journée peut par conséquent améliorer labsorption de la vitamine D.
Distribution et biotransformation
Elle est hydroxylée dans le foie pour former le 25-hydroxycholécalciférol, puis subi une nouvelle hydroxylation dans le rein pour former le métabolite actif 1,25-dihydroxycholécalciférol (calcitriol).
Elimination
Les métabolites circulent dans le sang sous forme liée à une α-globine. La vitamine D et ses métabolites sont principalement excrétés dans la bile et les fèces.
Particularités chez des groupes spécifiques de sujets ou de patients
La clairance métabolique rapportée chez les patients insuffisants rénaux est inférieur de 57 % à celle rapportée chez des volontaires sains.
Les sujets obèses ont plus de difficulté à maintenir leurs taux de vitamine D avec l'exposition au soleil et sont plus susceptibles de nécessiter des doses orales plus importantes de vitamine D pour pallier à leurs carences.
5.3. Données de sécurité préclinique
Dans des études de toxicité en administration répétée, les effets les plus fréquemment rapportés ont été les suivants : augmentation de la calciurie, diminution de la phosphaturie et de la protéinurie.
Une hypercalcémie a été rapportée à des doses élevées. En situation d'hypercalcémie prolongée, les altérations histologiques (calcification) ont été plus fréquemment observées au niveau des reins, du cur, de laorte, des testicules, du thymus et des muqueuses intestinales.
A fortes doses, le cholécalciférol sest avéré tératogène chez lanimal.
A des doses équivalentes aux doses thérapeutiques, le cholécalciférol na présenté aucune activité tératogène.
Le cholécalciférol ne présente aucune activité mutagène ou cancérigène potentielle.
Après première ouverture du flacon : le produit doit être conservé maximum 6 mois.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30 °C.
Ne pas congeler, ne pas mettre au réfrigérateur.
Conserver le flacon dans lemballage extérieur à labri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
Un embout compte-gouttes muni dune tige (en verre transparent) et dun bouchon (en polypropylène) est fourni. Chaque boîte contient 1 flacon et 1 embout compte-gouttes.
6.6. Précautions particulières délimination et de manipulation
Ne conserver aucun produit ou mélange alimentaire contenant DELTIUS pour une utilisation ultérieure ou un prochain repas (voir rubrique 4.2 Posologie et mode dadministration).
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
Contrada San Rafael, 3
POLig. INDustrial de ALCOBENDAS
28108 ALCOBENDAS (Madrid)
ESPAGNE
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[A compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[A compléter ultérieurement par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Médicament non soumis à prescription médicale.