RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 19/06/2017
LATANOPROST SANDOZ 50 microgrammes/ml, collyre en solution
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
1 ml de collyre contient 50 microgrammes de latanoprost (équivalent à 0,005 % m/v).
Une goutte contient environ 1,5 microgramme de latanoprost
Excipient à effet notoire : 1 ml contient 0,2 mg de chlorure de benzalkonium (équivalent à 0,02 % m/v).
Pour la liste complète des excipients, voir rubrique 6.1.
Solution incolore et limpide.
4.1. Indications thérapeutiques
Réduction de la pression intraoculaire élevée chez les patients pédiatriques atteints de pression intraoculaire élevée ou de glaucome pédiatrique.
4.2. Posologie et mode d'administration
Posologie recommandée chez les adultes (y compris le sujet âgé) :
La posologie recommandée est d'une goutte dans l'il (les yeux) atteint(s) une fois par jour.
L'effet optimal est obtenu quand LATANOPROST SANDOZ est administré le soir.
La posologie de latanoprost ne doit pas dépasser 1 instillation par jour, en effet, il a été montré qu'une fréquence d'administration supérieure diminue l'effet hypotenseur sur la pression intraoculaire.
En cas d'oubli, le traitement doit être poursuivi normalement, avec la dose suivante.
Comme pour tout collyre, afin de réduire une possible absorption systémique, une pression du sac lacrymal (occlusion ponctuelle) au niveau du canthus interne, pendant une minute, est recommandée après chaque instillation.
Les lentilles de contact doivent être retirées avant l'instillation du collyre et peuvent être remises 15 minutes après.
En cas d'utilisation concomitante de plusieurs collyres, les instillations de chacun des collyres doivent être espacées d'au moins cinq minutes.
Population pédiatrique
LATANOPROST SANDOZ peut être utilisé chez les enfants à la même posologie que chez l'adulte. Aucune donnée n'est disponible pour les nouveau-nés prématurés (moins de 36 semaines d'âge gestationnel). Les données dans le groupe d'âge <1 an (4 patients) sont très limitées (voir rubrique 5.1).
Mode dadministration
Voie ophtalmique.
Le flacon DROP-TAINER® est conçu pour assurer la délivrance d'une dose précise de médicament. Avant d'utiliser votre flacon DROP-TAINER®, lisez attentivement les instructions complètes.
1. Si vous utilisez d'autres médicaments ophtalmiques appliqués par voie topique, ils doivent être administrés au moins 5 minutes avant ou après LATANOPROST SANDOZ.
2. Lavez-vous les mains avant chaque utilisation.
3. Avant d'utiliser le médicament pour la première fois, vérifiez que le dispositif de sécurité du flacon est intact.
4. Tirez sur le dispositif de sécurité pour le briser.
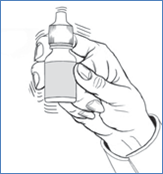
5. Avant chaque utilisation, agitez une fois le flacon et enlevez le bouchon à vis.
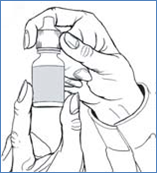
6. Renversez le flacon et maintenez-le entre le pouce et le majeur, avec les doigts pointant vers vous.

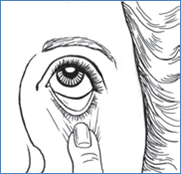
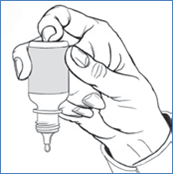
7. Inclinez votre tête en arrière et positionnez le flacon au-dessus de l'il affecté.
8. Avec la main opposée, placez un doigt sous l'il. Tirez légèrement vers le bas pour former une poche « en V » entre votre paupière inférieure et votre il. Ne touchez pas l'il avec le bout du flacon compte-gouttes.
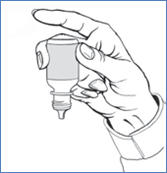
9. Avec la main tenant le flacon, placez votre index sur le fond du flacon. Appuyez sur le fond du flacon pour dispenser une goutte de médicament. Ne pressez pas les côtés du flacon. Gardez la tête inclinée vers l'arrière et fermez vos yeux pour permettre l'absorption du médicament dans l'il.
10. Répétez les étapes 6 à 9 pour lautre il si votre médecin vous la prescrit.
11. Replacez le bouchon à vis en tournant jusquà ce quil soit fermement en contact avec le flacon.
Hypersensibilité à la substance active ou à lun des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Ce changement de couleur des yeux a surtout été observé chez des patients ayant l'iris de plusieurs couleurs, c'est à dire bleu-marron, gris-marron, jaune-marron ou vert-marron.
Dans les essais cliniques avec le latanoprost, le début de ce changement de couleur des yeux survient en général dans les 8 premiers mois du traitement, rarement lors de la deuxième ou troisième année, et n'a pas été observé après la quatrième année de traitement.
Le taux de progression de la pigmentation irienne diminue avec le temps et est stable au bout de cinq ans. Les effets de l'augmentation pigmentaire au-delà de 5 ans n'ont pas été évalués. Dans un essai clinique en ouvert étudiant la tolérance du latanoprost sur cinq ans, 33 % des patients ont développé une pigmentation de l'iris (voir rubrique 4.8). La modification de la couleur de l'iris est discrète dans la majorité des cas et souvent, n'est pas observée cliniquement. Chez les patients ayant l'iris de plusieurs couleurs, l'incidence a été de 7 à 85 %, l'incidence la plus élevée ayant été observée chez les patients ayant l'iris de couleur jaune-marron.
Chez les patients ayant des yeux bleus de couleur uniforme, aucun changement de couleur n'a été observé et chez les patients ayant des yeux de couleur uniforme gris, verts ou marron, ces changements de couleur ont été rarement observés.
La modification de la couleur de l'iris est due à une augmentation de la teneur en mélanine des mélanocytes du stroma de l'iris et non à une augmentation du nombre de mélanocytes.
Généralement, la pigmentation brune entourant la pupille s'étend de façon concentrique vers la périphérie dans les yeux concernés, et l'iris peut devenir, totalement ou partiellement, brun plus foncé. Aucune augmentation ultérieure de la pigmentation brune de l'iris n'a été observée après l'arrêt du traitement. Cet effet n'a été associé à aucun symptôme ni modification pathologique dans les essais cliniques jusqu'à ce jour.
Ni les nævi, ni les éphélides de l'iris n'ont été affectés par le traitement. Aucune accumulation de pigment dans le trabeculum ou en d'autres points de la chambre antérieure n'a été observée lors des essais cliniques. Sur la base de 5 années d'expérience clinique, l'augmentation de la pigmentation irienne n'a entraîné aucune séquelle clinique néfaste et le latanoprost peut être poursuivi en cas de pigmentation irienne. Cependant, les patients devront être suivis régulièrement et si le contexte clinique l'impose, le traitement par latanoprost pourra être arrêté.
L'expérience du latanoprost est limitée dans le glaucome chronique à angle fermé, dans le glaucome à angle ouvert des patients pseudophaques et dans le glaucome pigmentaire. Il n'y a pas d'expérience du latanoprost dans les glaucomes inflammatoires et néovasculaires ou dans des conditions d'inflammation oculaire.
Le latanoprost a peu ou pas d'effet sur la pupille mais il n'a pas été expérimenté dans les crises de glaucome aigu par fermeture de l'angle. Il est donc recommandé d'utiliser la latanoprost avec précaution, dans ces conditions, tant que les connaissances ne sont pas plus approfondies.
Il y a peu de données cliniques sur l'utilisation du latanoprost pendant la période péri-opératoire d'une chirurgie de la cataracte. Le latanoprost doit être utilisé avec précaution chez ces patients.
Le latanoprost doit être utilisé avec précaution chez les patients ayant des antécédents de kératite herpétique, et doit être évité en cas de kératite à herpes simplex active et chez les patients ayant des antécédents de kératite herpétique récurrente associée aux analogues des prostaglandines.
Des cas d'dèmes maculaires ont été rapportés (voir rubrique 4.8) principalement chez des patients aphaques, chez des patients pseudophaques présentant une rupture capsulaire postérieure ou porteurs d'un implant en chambre antérieure ou chez des patients ayant des facteurs de risque connus d'dème maculaire cystoïde (tels que les rétinopathies diabétiques et les occlusions veineuses rétiniennes). Le latanoprost doit être utilisé avec précaution chez les patients aphaques, chez les patients pseudophaques présentant une rupture capsulaire postérieure ou porteurs d'implant en chambre antérieure ainsi que chez les patients ayant des facteurs de risque connus d'dème maculaire cystoïde.
Chez les patients présentant des facteurs de risques connus d'iritis/uvéites, le latanoprost devra être utilisé avec précaution.
L'expérience du latanoprost chez les patients asthmatiques est limitée, toutefois des cas d'aggravation d'asthme et/ou de dyspnée ont été rapportés après commercialisation. Le latanoprost doit être utilisé avec précaution chez les patients asthmatiques jusqu'à ce que l'expérience soit suffisante (voir également rubrique 4.8).
Une modification de la coloration de la peau périorbitaire a été observée, la majorité des cas rapportés concernant des patients Japonais. A ce jour, l'expérience montre que cette coloration de la peau périorbitaire n'est pas définitive et même, que dans certains cas, elle est réversible alors que le traitement par latanoprost est poursuivi.
Le latanoprost peut progressivement modifier les cils et le duvet palpébral de l'il traité et de ses contours.
Ces changements incluent des cils ou un duvet plus long, plus épais, plus foncés, en nombre plus important et une pousse mal orientée des cils. Les changements au niveau des cils sont réversibles à l'arrêt du traitement.
LATANOPROST SANDOZ contient le conservateur chlorure de benzalkonium (0,2 mg/ml), qui peut être absorbé par les lentilles de contact souples et peut les décolorer.
Les lentilles de contact doivent être retirées avant l'instillation et peuvent être remises 15 minutes après l'administration.
Le chlorure de benzalkonium peut entraîner une irritation des yeux, les yeux secs et peut affecter la surface de la cornée.
LATANOPROST SANDOZ doit être utilisé avec prudence chez les patients présentant une sécheresse oculaire et chez les patients présentant une atteinte cornéenne. Par ailleurs, une surveillance est nécessaire en cas dutilisation prolongée chez de tels patients.
Population pédiatrique
Les données d'efficacité et de sécurité dans le groupe d'âge <1 an (4 patients) sont très limitées (voir rubrique 5.1). Aucune donnée n'est disponible pour les nouveau-nés prématurés (moins de 36 semaines d'âge gestationnel).
Chez les enfants de 0 à moins de 3 ans qui souffrent surtout de GCP (glaucome primaire congénital), la chirurgie (trabéculectomie par exemple / goniotomie) reste le traitement de première ligne.
La sécurité à long terme chez les enfants n'a pas encore été établie.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucun résultat conclusif d'interaction de LATANOPROST SANDOZ avec d'autres médicaments n'est disponible à ce jour.
Des élévations paradoxales de la pression intraoculaire ont été rapportées suite à l'administration ophtalmique concomitante de deux analogues de prostaglandines. Par conséquent, l'utilisation de deux ou plus de deux prostaglandines, analogues de prostaglandine, ou dérivés de prostaglandine n'est pas recommandée.
Population pédiatrique
Les études d'interaction n'ont été réalisées que chez l'adulte.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'y a pas de donnée de sécurité concernant l'utilisation de cette spécialité pendant la grossesse. Elle possède des effets pharmacologiques potentiellement dangereux sur le déroulement de la grossesse, le ftus ou le nouveau-né. Par conséquent, le latanoprost ne doit pas être utilisé au cours de la grossesse.
Allaitement
Le latanoprost et ses métabolites peuvent passer dans le lait maternel et par conséquent, le latanoprost ne doit pas être utilisé chez les femmes qui allaitent ou bien, l'allaitement doit être interrompu.
Fertilité
Aucun effet du latanoprost sur la fertilité mâle ou femelle na été observé lors des études chez lanimal (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Dans ce cas, les patients ne doivent pas conduire ni utiliser de machines jusqu'à ce que la vision redevienne normale.
La majorité des effets indésirables se rapporte au système oculaire. Dans un essai clinique en ouvert étudiant la tolérance du latanoprost sur 5 ans, 33 % des patients ont développé une augmentation de la pigmentation irienne (voir rubrique 4.4). D'autres effets indésirables oculaires sont généralement passagers et surviennent à l'administration de la dose.
Les effets indésirables sont classés selon leur fréquence d'apparition, comme suit : très fréquents (≥ 1/10), fréquents (≥ 1/100 à <1/10), peu fréquents (≥ 1/1000 à <1/100), rares (≥ 1/10000 à <1/1000) et très rares (<1/10000). Fréquence indéterminée : ne peut être estimée sur la base des données disponibles.
|
Classe Système Organe |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Très rare |
Fréquence indéterminée |
|
Infections et infestations |
|
|
|
|
|
Kératite herpétique |
|
Affections du système nerveux |
|
|
|
|
|
Céphalées, étourdissement |
|
Affections oculaires |
Augmentation de la pigmentation de l'iris, hyperhémie conjonctivale légère ou modérée, irritation oculaire (sensation de brûlure, grain de sable, démangeaison, picotement et sensation de corps étranger), modifications des cils et du duvet palpébral (augmentation de la longueur, de l'épaisseur, de la pigmentation et du nombre) (cas majoritairement recensés dans la population japonaise) |
Kératites ponctuées superficielles transitoires, le plus souvent asymptomatiques, blépharite, douleur oculaire |
dème palpébral, sécheresse oculaire, kératite, vision trouble, conjonctivite |
Iritis/uvéite (cas majoritairement rapportés chez des patients présentant des facteurs de risque prédisposant associés), dème maculaire, dème cornéen et ulcérations cornéennes symptomatiques, dème péri-orbitaire, cils mal orientés engendrant parfois une irritation oculaire, rangée supplémentaire de cils au niveau de l'ouverture des glandes de Meibomius (distichiasis), photophobie |
Modifications périorbitaires et palpébrales se traduisant par un creusement du sillon palpébral |
Kyste irien |
|
Affections cardiaques |
|
|
|
|
Aggravation de l'angine de poitrine chez des patients présentant une pathologie angineuse pré-existante |
Palpitations |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
|
Asthme, exacerbation de l'asthme et dyspnée |
|
|
|
Affections de la peau et du tissu sous-cutané |
|
|
Eruptions cutanées |
Réaction cutanée locale au niveau des paupières, coloration plus foncée des paupières |
|
|
|
Affections musculo-squelettiques et systémiques |
|
|
|
|
|
Myalgie, arthralgie. |
|
Troubles généraux et anomalies au site d'administration |
|
|
|
|
Douleurs thoraciques. Des cas de calcification cornéenne ont été très rarement rapportés en association avec l'utilisation de collyres contenant des phosphates chez certains patients dont la cornée était endommagée de façon importante. |
|
Population pédiatrique
Dans deux essais cliniques à court terme (≤ 12 semaines) impliquant 93 (25 et 68) patients pédiatriques le profil d'innocuité était semblable à celui des adultes et aucun nouvel effet indésirable n'a été identifié. Les profils de sécurité à court terme dans les différents sous-ensembles pédiatriques étaient également similaires (voir rubrique 5.1). Les effets indésirables observés le plus souvent dans la population pédiatrique par rapport aux adultes sont les suivants: rhinopharyngite et pyrexie.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
En cas d'ingestion accidentelle de LATANOPROST SANDOZ, les informations suivantes peuvent être utiles : un flacon contient 125 microgrammes de latanoprost. Plus de 90 % du principe actif est métabolisé lors du premier passage hépatique. L'injection intraveineuse de latanoprost chez les volontaires sains à la dose de 3 microgrammes/kg a donné des concentrations plasmatiques 200 fois supérieures à celles observées au cours du traitement clinique et n'a entraîné l'apparition d'aucun symptôme, mais une dose comprise entre 5,5 et 10 microgrammes/kg a provoqué des nausées, des douleurs abdominales, des étourdissements, une fatigue, des bouffées de chaleur et des sueurs. Chez le singe, le latanoprost a été injecté par voie intraveineuse à des doses allant jusqu'à 500 microgrammes/kg, sans effet notable sur le système cardio-vasculaire.
L'administration intraveineuse de latanoprost chez le singe a été associée à une bronchoconstriction transitoire. Aucune bronchoconstriction n'a toutefois été observée chez des patients présentant un asthme modéré après l'instillation du latanoprost, à une dose égale à sept fois la dose thérapeutique recommandée pour latanoprost.
En cas de surdosage de latanoprost, le traitement devra être symptomatique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Le principe actif, le latanoprost, analogue de la prostaglandine F2α, est un agoniste sélectif des récepteurs FP aux prostanoïdes qui abaisse la pression intraoculaire en augmentant l'écoulement de l'humeur aqueuse. Chez l'homme, la diminution de la pression intraoculaire débute environ trois à quatre heures après l'administration de latanoprost, et l'effet maximum est observé au bout de huit à douze heures. La réduction de la pression est maintenue pendant au moins 24 heures.
Des études conduites chez les animaux et chez l'homme indiquent que le latanoprost agit principalement en augmentant l'écoulement par la voie uvéosclérale, même s'il a été rapporté chez l'homme une augmentation de la facilité d'écoulement par les voies usuelles (diminution de la résistance).
Des études pivots ont montré l'efficacité de latanoprost en monothérapie. De plus, des études cliniques évaluant l'utilisation de latanoprost en association ont été conduites. Elles comprennent des études montrant que le latanoprost est efficace en association avec les bêta-bloquants (timolol). Des études à court terme (1 ou 2 semaines) suggèrent un effet additif du latanoprost administré en association avec des collyres sympathomimétiques (dipivéphrine), des inhibiteurs de l'anhydrase carbonique par voie orale (acétazolamide) et un effet au moins partiellement additif avec des collyres parasympathomimétiques (pilocarpine).
Des essais cliniques ont montré que le latanoprost n'a pas d'effet significatif sur la production d'humeur aqueuse. En outre, aucune action sur la barrière hémato-aqueuse n'a été observée.
Chez le singe, l'effet du latanoprost sur la circulation sanguine intraoculaire est nul ou négligeable après administration à la dose thérapeutique. Néanmoins, une hyperhémie conjonctivale ou épisclérale légère à modérée peut être observée lors d'un traitement local.
Après un traitement chronique par le latanoprost chez le singe ayant auparavant subi une extraction du cristallin extracapsulaire, aucune lésion des vaisseaux sanguins rétiniens n'a été mise en évidence par l'angiographie à la fluorescéine.
Un traitement à court terme par le latanoprost n'a pas induit de fuite de la fluorescéine dans le segment postérieur des yeux humains pseudophaques.
Administré aux doses thérapeutiques, le latanoprost n'a été associé à aucun effet pharmacologique significatif sur l'appareil cardio-vasculaire ou respiratoire.
Population pédiatrique
L'efficacité du latanoprost chez des patients de ≤ 18 ans a été démontrée dans une étude clinique en double aveugle d'une durée de 12 semaines comparant le latanoprost au timolol chez 107 patients diagnostiqués avec une hypertension oculaire et un glaucome pédiatrique. Les nouveau-nés devaient avoir un âge gestationnel d'au moins 36 semaines. Les patients étaient randomisés soit avec du latanoprost 50 microgrammes/ml une fois par jour soit avec du timolol 0,5 % (ou optionnellement du timolol 0,25 % pour les patients âgés de moins de 3 ans) deux fois par jour. Le critère primaire d'efficacité était la réduction moyenne de la pression intraoculaire initiale après 12 semaines de traitement. La réduction moyenne de la pression intraoculaire était similaire dans les 2 groupes traités (latanoprost et timolol). Dans toutes les tranches d'âge étudiées (0 à <3 ans, 3 à <12 ans puis entre 12 et 18 ans), la réduction moyenne de la pression intraoculaire après 12 semaines de traitement dans le groupe latanoprost restait comparable à celle du groupe timolol. Toutefois, les données d'efficacité dans le groupe latanoprost pour la tranche d'âge 0 à <3 ans n'ont été recueillies que pour 13 patients et aucune efficacité pertinente n'a été observée chez les 4 patients dont l'âge était <1 an dans l'étude clinique pédiatrique. Aucune donnée n'est disponible chez les enfants nés avant terme (âge gestationnel inférieur à 36 semaines).
De même, la réduction de pression intraoculaire parmi les sujets du sous-groupe souffrant de glaucome congénital/ infantile (PCG) était similaire dans les 2 groupes traités (latanoprost et timolol). Des résultats comparables ont été observés dans l'autre sous-groupe (Non-PCG, ex: glaucome juvénile à angle ouvert, glaucome aphaque).
L'effet du traitement sur la pression intraoculaire a été observé après la première semaine de traitement et s'est poursuivi durant les 12 semaines de l'étude, comme chez l'adulte.
|
Tableau: Réduction de la pression intraoculaire (mmHg) après 12 semaines en fonction du groupe de traitement actif et du diagnostic au début du traitement |
|
||||
|
|
Latanoprost |
Timolol |
|
||
|
Moyenne à l'inclusion (SE) |
27,3 (0,75) |
27,8 (0,84) |
|
||
|
Variation moyenne par rapport à la valeur initiale après 12 semaines de traitement (SE) |
-7,18 (0,81) |
-5,72 (0,81) |
|
||
|
p-value vs. timolol |
0,2056 |
|
|||
|
|
PCG |
Non-PCG |
PCG |
Non-PCG |
|
|
Moyenne à l'inclusion (SE) |
26,5 (0,72) |
28,2 (1,37) |
26,3 (0,95) |
29,1 (1,33) |
|
|
Variation moyenne par rapport à la valeur initiale après 12 semaines de traitement (SE) |
-5,90 (0,98) |
-8,66 (1,25) |
-5,34 (1,02) |
-6,02 (1,18) |
|
|
p-value vs. timolol |
0,6957 |
0,1317 |
|
|
|
SE : écart à la moyenne,
Moyenne ajustée basée sur le modèle d'analyse de covariance (ANCOVA).
5.2. Propriétés pharmacocinétiques
La prodrogue est bien absorbée par la cornée et la totalité de la substance pénétrant dans l'humeur aqueuse est hydrolysée au cours du passage à travers la cornée.
Les études conduites chez l'homme ont montré que le pic de concentration dans l'humeur aqueuse est atteint environ deux heures après administration locale. Après application locale chez le singe, le latanoprost est principalement distribué dans le segment antérieur, la conjonctive et les paupières. Seule une quantité infime atteint le segment postérieur.
L'acide de latanoprost n'est pratiquement pas métabolisé dans l'il. Le métabolisme est principalement hépatique. Sa demi-vie plasmatique chez l'homme est de 17 minutes. Les études animales ont montré une activité faible, voire nulle, des métabolites de l'acide de latanoprost, le 1, 2 - dinor et le 1, 2, 3, 4 - tétranor, qui sont principalement éliminés dans l'urine.
Population pédiatrique
Une étude de pharmacocinétique en ouvert sur les concentrations plasmatiques d'acide de latanoprost a été menée chez 22 adultes et 25 patients pédiatriques (de la naissance à l'âge de <18 ans) souffrant d'hypertension oculaire et de glaucome. Toutes les tranches d'âge ont été traitées avec du latanoprost 50 microgrammes/ml, une goutte par jour dans chaque il pendant au minimum 2 semaines. L'exposition systémique à l'acide de latanoprost était environ 2 fois plus importante chez les enfants âgés de 3 à <12 ans et 6 fois plus importante chez les enfants âgés de moins de 3 ans que chez les adultes, mais une large marge de sécurité pour la survenue d'effets indésirables systémiques était maintenue (voir rubrique 4.9). La durée moyenne d'atteinte du pic de concentration plasmatique était de 5 minutes après l'administration de la dose dans toutes les tranches d'âge.
La demi-vie moyenne d'élimination plasmatique était courte (< 20 minutes) et du même ordre chez les patients enfants et adultes, n'entraînant pas d'accumulation d'acide de latanoprost dans la circulation systémique à l'état d'équilibre.
5.3. Données de sécurité préclinique
Celle-ci est probablement due à une bronchoconstriction de courte durée. Les études animales n'ont pas révélé d'action sensibilisante du latanoprost.
Aucun effet toxique n'a été détecté dans l'il à des doses allant jusqu'à 100 microgrammes/il/jour chez le lapin ou le singe (la dose thérapeutique est environ 1,5 microgrammes/il/jour). Chez le singe, toutefois, il a été montré que le latanoprost induisait une augmentation de la pigmentation de l'iris.
Le mécanisme conduisant à une augmentation de la pigmentation semble être une stimulation de la production de mélanine dans les mélanocytes de l'iris, sans prolifération des mélanocytes. La modification de la couleur de l'iris peut être permanente.
Lors des études de toxicité oculaire chronique, le latanoprost administré à la dose de 6 microgrammes/il/jour a été associé à une augmentation de la fente palpébrale. Cette action est réversible et se produit pour des doses supérieures aux doses thérapeutiques. Elle n'a pas été observée chez l'homme.
Les tests de mutation reverse sur des bactéries, les tests de mutation génique sur le lymphome de souris, ainsi que le test du micronoyau chez la souris, se sont révélés négatifs avec le latanoprost. Des aberrations chromosomiques ont été observées in vitro sur des lymphocytes humains. Des effets similaires ont été notés avec une prostaglandine naturelle, la prostaglandine F2a, ce qui indique un effet de classe.
D'autres études de mutagénèse, concernant la synthèse non programmée d'ADN in vitro/in vivo chez le rat, ont été négatives et montrent que le latanoprost n'a pas d'action mutagène.
Les études de carcinogénèse chez la souris et le rat ont également été négatives.
Aucun effet du latanoprost sur la fertilité mâle ou femelle n'a été observé lors des études chez l'animal. Dans l'étude d'embryotoxicité chez le rat, aucun effet embryotoxique n'a été montré après administration intraveineuse de latanoprost (aux doses de 5, 50 et 250 microgrammes/kg/jour). Toutefois, le latanoprost a induit un effet embryon-létal chez le lapin à des doses égales ou supérieures à 5 microgrammes/kg/jour.
La dose de 5 microgrammes/kg/jour (environ 100 fois la dose thérapeutique) a entraîné une toxicité embryo-ftale significative, caractérisée par une incidence accrue des résorptions tardives, des avortements, ainsi que par une diminution du poids ftal.
Aucun pouvoir tératogène n'a été détecté.
Après 1ère ouverture du flacon : 4 semaines.
6.4. Précautions particulières de conservation
A conserver au réfrigérateur (entre 2°C et 8°C).
Conserver le flacon dans l'emballage extérieur, à l'abri de la lumière.
Après 1ère ouverture du flacon : A conserver à une température ne dépassant pas 25°C.
6.5. Nature et contenu de l'emballage extérieur
2,5 ml en flacon DROP-TAINER® en PEBD naturel de 4 ml muni d'un embout compte-gouttes en PEBD naturel, d'un bouchon à vis turquoise en polypropylène (PP) et dune bande protectrice en PVC autour du col et du bouchon.
Chaque flacon compte-gouttes contient 2,5 ml de solution correspondant approximativement à 90 gouttes.
Boîtes de 1, 3 et 6 flacons de 2,5 ml.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Pas d'exigences particulières.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
49 AVENUE GEORGES POMPIDOU
92300 LEVALLOIS-PERRET
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 219 375 3 4 : 2,5 ml en flacon compte-gouttes (PEBD). Boîte de 1 flacon.
· 34009 219 377 6 3 : 2,5 ml en flacon compte-gouttes (PEBD). Boîte de 3 flacons.
· 34009 219 378 2 4 : 2,5 ml en flacon compte-gouttes (PEBD). Boîte de 6 flacons.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[A compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[A compléter ultérieurement par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I