|
|
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 12/09/2017
XEOMIN 50 unités, poudre pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
1 flacon contient 50 unités de neurotoxine de Clostridium botulinum de type A (150 kD), sans protéines complexantes*.
* Neurotoxine botulinique de type A purifiée obtenue par culture de Clostridium botulinum (souche Hall).
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour solution injectable.
Poudre blanche.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Les tests évaluant lactivité biologique de la neurotoxine sont différents. Aussi, les unités de XEOMIN ne sont pas interchangeables avec les autres préparations de toxine botulinique de type A.
Pour une information plus complète sur les études cliniques comparant XEOMIN à la toxine botulinique de type A conventionnelle (900kD), voir rubrique 5.1.
Cas général
XEOMIN doit être administré uniquement par des médecins ayant les qualifications adéquates et une bonne expérience de l'utilisation de la toxine botulinique de type A.
Les doses optimales, la fréquence et le nombre de sites d'injection dans le muscle traité doivent être déterminés par le médecin, individuellement pour chaque patient. La dose optimale doit être déterminée par augmentation progressive des doses.
Posologie
Blépharospasme
La dose initiale recommandée est de 1,25 à 2,5 unités par site dinjection. La dose initiale ne doit pas dépasser 25 unités par il. La dose totale ne doit pas dépasser 100 unités toutes les 12 semaines. Les intervalles de traitement par XEOMIN doivent être adaptés aux besoins cliniques réels et individuels du patient.
L'effet du traitement apparaît dans un délai médian de quatre jours suivant l'injection. Généralement, leffet du traitement par XEOMIN dure environ 3 à 4 mois ; il peut toutefois durer plus ou moins longtemps. Le traitement peut être répété si nécessaire.
Si le résultat du traitement initial est considéré comme insuffisant, la dose peut être au maximum doublée à la session suivante. Cependant, l'injection de plus de 5 unités par site ne semble apporter aucun bénéfice supplémentaire.
Torticolis spasmodique
Pour le traitement du torticolis spasmodique, la dose de XEOMIN doit être adaptée individuellement pour chaque patient selon l'orientation de la tête et du cou du patient, la localisation de la douleur éventuelle, l'hypertrophie du muscle, le poids du patient et sa réponse à linjection.
Au cours de la première session de traitement, la dose maximale de 200 unités doit être respectée. En fonction de la réponse, des ajustements posologiques peuvent être réalisés au cours des sessions suivantes. Ne pas administrer plus de 300 unités au cours dune séance. Ne pas administrer plus de 50 unités par site dinjection.
L'effet du traitement apparaît dans un délai médian de sept jours suivant l'injection. Généralement, l'effet de XEOMIN dure environ 3 à 4 mois ; il peut toutefois durer plus ou moins longtemps. Bien que des intervalles de traitement de moins de 10 semaines ne soient pas recommandés, les intervalles de traitement par XEOMIN doivent être adaptés aux besoins cliniques réels et individuels du patient.
Spasticité des membres supérieurs
La posologie exacte et le nombre de sites dinjection doivent être adaptés à chaque patient d'après la taille, le nombre et l'emplacement des muscles impliqués, la sévérité de la spasticité et la présence d'une faiblesse musculaire locale.
Doses recommandées par muscle :
|
Tableau clinique Muscle |
Unités (Gamme) |
Nombre de sites dinjection par muscle |
|
Fléchisseurs du poignet |
||
|
Flexor carpi radialis |
25-100 |
1-2 |
|
Flexor carpi ulnaris |
20-100 |
1-2 |
|
Fléchisseurs des doigts |
||
|
Flexor digitorum superficialis |
25-100 |
2 |
|
Flexor digitorum profundus |
25-100 |
2 |
|
Fléchisseurs du coude |
||
|
Brachioradialis |
25-100 |
1-3 |
|
Biceps |
50-200 |
1-4 |
|
Brachialis |
25-100 |
1-2 |
|
Pronateurs de lavant-bras |
||
|
Pronator quadratus |
10-50 |
1 |
|
Pronator teres |
25-75 |
1-2 |
|
Fléchisseurs propres du pouce |
||
|
Flexor pollicis longus |
10-50 |
1 |
|
Adductor pollicis |
5-30 |
1 |
|
Flexor pollicis brevis/Opponens pollicis |
5-30 |
1 |
|
Rotateurs internes, extenseurs et adducteurs de lépaule |
|
|
|
Deltoideus, pars clavicularis |
20-150 |
1-3 |
|
Latissimus dorsi |
25-150 |
1-4 |
|
Pectoralis major |
20-200 |
1-6 |
|
Subscapularis |
15-100 |
1-4 |
|
Teres major |
20-100 |
1-2 |
La dose totale maximale par session de traitement de la spasticité des membres supérieurs ne doit pas excéder 500 unités et pas plus de 250 unités dans les muscles de lépaule.
Les patients ont noté une efficacité clinique dans les 4 jours avec une amélioration maximale du tonus musculaire dans les 4 semaines qui suivent linjection. Comme leffet du traitement persiste, en général, plus ou moins 3 mois ; les réinjections peuvent être espacées dau moins 12 semaines. De ce fait, les intervalles de traitement par XEOMIN doivent être adaptés aux besoins cliniques réels et individuels du patient.
Toutes les indications
En l'absence d'amélioration dans le mois qui suit la première séance d'injections, il y a lieu de :
· Vérifier cliniquement leffet de la toxine sur le muscle injecté (ex : un examen électromyographique en milieu spécialisé).
· Analyser les causes de non-réponse telles que : une mauvaise isolation des muscles injectés, une dose insuffisante, une technique d'injection inadaptée, une contracture fixe, des muscles antagonistes trop faibles, la formation éventuelle d'anticorps.
· Réévaluer la pertinence dun traitement par neurotoxine botulinique de type A.
· En l'absence d'effets indésirables lors du traitement initial, une deuxième session dinjections peut être réalisée dans les conditions suivantes :
1) ajustement de la dose, en tenant compte de l'échec précédent ;
2) localisation du muscle à injecter à laide de techniques telles que le guidage électromyographique ;
3) respect des intervalles de temps minimum entre les injections.
La tolérance et lefficacité de XEOMIN chez les enfants et les adolescents de la naissance à 17 ans na pas encore été établie. XEOMIN n'est donc pas recommandé dans cette population pédiatrique jusqu'à l'obtention de données supplémentaires.
Mode dadministration
Toutes les indications
Pour les instructions sur la reconstitution des flacons avant administration et les instructions délimination des flacons, voir rubrique 6.6. Après reconstitution, XEOMIN doit être utilisé pour une seule session d'injection et pour un seul patient.
La solution reconstituée de XEOMIN est destinée à la voie intramusculaire.
Blépharospasme
La solution reconstituée de XEOMIN est injectée au moyen d'une aiguille stérile appropriée (diamètre de 27-30 Gauge/0,30-0,40 mm et longueur de 12,5 mm). Le guidage électromyographique n'est pas nécessaire. Un volume d'injection d'environ 0,05 à 0,1 ml est recommandé.
XEOMIN est injecté dans la partie interne et externe du muscle orbiculaire de la paupière supérieure et dans la partie externe latérale du muscle orbiculaire de la paupière inférieure. Si des spasmes gênent la vision, le produit peut être injecté dans l'arcade sourcilière, la partie latérale du muscle orbiculaire et la zone supérieure du visage.
Torticolis spasmodique
Une aiguille stérile appropriée (diamètre de 25-30 Gauge/0,30-0,50 mm et longueur de 37 mm) doit être utilisée pour l'injection dans les muscles superficiels et par exemple, une aiguille de diamètre de 22 Gauge/0,70 mm et de longueur de 75 mm pour des injections dans les muscles plus profonds. Un volume d'injection d'environ 0,1 à 0,5 ml par site dinjection est recommandé.
Pour le traitement du torticolis spasmodique, XEOMIN est injecté dans le sterno-cléido-mastoïdien, le muscle élévateur de l'omoplate, les scalènes, le splénius et/ou les trapèzes. Cette liste n'est pas exhaustive car tous les muscles contrôlant la position de la tête peuvent être concernés et nécessiter un traitement. En cas de difficulté pour isoler les muscles, les injections peuvent être réalisées à laide de techniques telles que le guidage électromyographique ou échographique. La masse musculaire et le degré d'hypertrophie ou d'atrophie sont des facteurs à prendre en considération dans le choix de la dose.
La multiplicité des sites d'injection permet une diffusion plus uniforme de XEOMIN dans les zones innervées du muscle dystonique et s'avère particulièrement utile dans les grands muscles. Le nombre optimal de sites dinjection dépend de la taille des muscles à dénerver chimiquement.
Le sterno-cléido-mastoïdien ne doit pas être injecté de façon bilatérale ni recevoir des doses supérieures à 100 U, en raison dun risque accru d'événements indésirables (en particulier de dysphagie).
Spasticité des membres supérieurs
La solution reconstituée de XEOMIN est injectée au moyen d'une aiguille stérile appropriée (ex : 26 Gauge/0,45 mm de diamètre/37 mm de longueur, pour les muscles superficiels et une aiguille plus longue ex : 22 Gauge/0,7 mm de diamètre/75 mm de longueur, pour les muscles plus profonds).
La localisation des muscles à laide de techniques telles que le guidage électromyographique ou échographique est recommandée en cas de difficulté pour isoler un muscle. La multiplicité des sites dinjection permet un contact plus uniforme de XEOMIN avec les zones innervées des muscles et s'avère particulièrement utile pour les grands muscles.
· Hypersensibilité à la substance active ou à l'un des excipients mentionés à la rubrique 6.1.
· Troubles généralisés de l'activité musculaire (ex: myasthénie grave, syndrome de Lambert-Eaton).
· Infection ou inflammation au site d'injection concerné.
4.4. Mises en garde spéciales et précautions d'emploi
Avant toute administration de XEOMIN, le médecindoit se familiariser à lanatomie du patient et à toute modification anatomique consécutive à des traitements chirurgicaux antérieurs.
Des précautions doivent être prises afin de sassurer que XEOMIN nest pas injecté dans un vaisseau sanguin. Pour le traitement de la dystonie cervicale et de la spasticité, XEOMIN doit être injecté avec précaution au niveau des sites situés à proximité de zones sensibles tels que la carotide, lapex des poumons et lsophage.
XEOMIN doit être utilisé avec précaution :
· en cas de troubles hémorragiques
· chez les patients sous traitement anticoagulant ou sous un traitement susceptible davoir un effet anticoagulant.
Ne pas dépasser les doses individuelles recommandées.
Il convient de rappeler aux patients précédemment sédentaires ou akinétiques que la reprise d'activité après une injection de XEOMIN doit être progressive.
Les effets cliniques de la neurotoxine botulinique de type A peuvent varier aux cours des sessions dinjections. Les raisons de ces changements peuvent être en relation avec la technique de reconstitution, les intervalles de traitement choisis, les muscles injectés ou lactivité de la toxine qui peut légèrement différer selon la procédure de dosage de lactivité biologique choisie ou une non-réponse secondaire.
Diffusion locale ou à distance de la toxine
Des effets indésirables, liés à une injection de toxine botulinique de type A mal ciblée peut temporairement paralyser les muscles situés à proximité. De fortes doses peuvent causer une paralysie des muscles situés à distance du site dinjection.
Des effets indésirables liés à la diffusion de la toxine botulinique de type A à distance du site d'administration, ont été rapportés (voir rubrique 4.8). Des cas de mise en jeu du pronostic vital et de décès lesquels dans certains cas, étaient associés à une dysphagie, une pneumonie et/ou une faiblesse extrême, ont été rapportés.
Des cas de dysphagie ont également été rapportés à la suite dinjections dans dautres sites que les muscles du cou.
Troubles musculaires préexistants
Les patients traités à dose thérapeutique peuvent présenter une faiblesse musculaire excessive. Les patients souffrant de troubles neuromusculaires sont plus à risque de présenter cet effet. Chez ces patients,la toxine botulinique de type A devra être utilisée sous le contrôle d'un médecin spécialiste et uniquement si le bénéfice du traitement l'emporte sur le risque. Les patients ayant des antécédents de dysphagie et de pneumopathie d'inhalation doivent être traités avec la plus grande précaution.
Les patients et leur entourage doivent être avertis de la nécessité d'une prise en charge médicale immédiate en cas de troubles de la déglutition, de troubles de lélocution ou de troubles respiratoires.
XEOMIN doit être utilisé avec précaution :
· Chez les patients souffrant de sclérose latérale amyotrophique
· Chez les patients présentant d'autres troubles entraînant un dysfonctionnement neuromusculaire périphérique,
· Dans les muscles cibles qui présentent une faiblesse ou une atrophie prononcée.
Réactions dhypersensibilité
Des réactions dhypersensibilité ont été rapportées avec la neurotoxine botulinique de type A. En cas de réaction dhypersensibilité grave (ex : réaction anaphylactique) et/ou immédiate, un traitement médical approprié doit être instauré.
Formation danticorps
Des doses trop fréquentes peuvent augmenter le risque de formation d'anticorps et conduire à un échec thérapeutique (voir rubrique 4.2).
La formation potentielle danticorps peut être minimisée par ladministration de la plus petite dose efficace indiquée et ce, dans le respect des intervalles dinjection minimum entre 2 sessions.
Indications
Blépharospasme
Des injections à proximité du muscle releveur de la paupière supérieure doivent être évitées afin de réduire la survenue dune ptose. Une diplopie peut survenir en cas de diffusion de la neurotoxine botulinique dans le muscle oblique inférieur. Eviter dinjecter dans la partie médiane de la paupière inférieure pour réduire ce risque.
Du fait de l'activité anticholinergique de la neurotoxine botulinique de type A, XEOMIN doit être utilisé avec précaution chez les patients à risque de développer un glaucome par fermeture de l'angle.
Afin de prévenir l'apparition d'un ectropion, des injections dans la paupière inférieure doivent être évitées et, si besoin, un traitement vigoureux en cas de lésion épithéliale doit être initié à base de collyre protecteur, de pommade, de lentilles souples thérapeutiques ou par occlusion de l'il à laide dun bandeau ou de tout autre moyen similaire.
La diminution du clignement due à l'injection de XEOMIN dans le muscle orbiculaire peut conduire à une exposition prolongée de la cornée, à une lésion épithéliale persistante et à une ulcération de la cornée, en particulier chez les patients ayant présenté des troubles du nerf crânien (nerf facial). Des tests de sensibilité cornéenne doivent être effectués chez les patients ayant été opérés des yeux.
Des ecchymoses se produisent facilement dans les tissus mous de la paupière. L'application d'une légère pression immédiatement après l'injection peut limiter ce risque.
Torticolis spasmodique
Les patients doivent être informés que les injections de XEOMIN pour le traitement du torticolis spasmodique peuvent provoquer une dysphagie légère à sévère avec un risque de fausses routes et de dyspnée. Des mesures peuvent s'avérer nécessaires comme par exemple la pose dune sonde d'alimentation gastrique (voir rubrique 4.8). Le risque de survenue dune dysphagie est minimisé en limitant la dose dinjection à 100 unités maximum dans le muscle sterno-cléido-mastoïdien. Les patients ayant une faible masse musculaire au niveau du cou ou qui nécessitent des injections bilatérales dans le muscle sterno-cléido-mastoïdien présentent des risques plus élevés. L'apparition d'une dysphagie est liée à l'effet pharmacologique de XEOMIN suite à la diffusion de la neurotoxine dans la musculature sophagienne.
Spasticité des membres supérieurs
XEOMIN comme traitement local de la spasticité, a été étudié en association avec les traitements habituels et ne vise pas à les remplacer. XEOMIN n'est pas efficace dans l'amélioration des mouvements au niveau d'une articulation touchée par une contracture fixe.
Lapparition ou la récurrence de crises convulsives ont été rapportées chez les patients prédisposés. Limputabilité à la toxine botulinique de type A na pas été établie.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude dinteractions médicamenteuses na été réalisée.
Théoriquement, l'effet de la neurotoxine botulinique peut être potentialisé par les aminosides ou par d'autres médicaments qui interfèrent avec la transmission neuromusculaire, tels que les myorelaxants de type tubocurarine.
Par conséquent, l'utilisation concomitante de XEOMIN avec des aminosides ou la spectinomycine nécessite une vigilance particulière. Les myorelaxants périphériques sont à utiliser avec précaution ; au besoin en réduisant la dose de départ du myorelaxant ou en utilisant une substance à action intermédiaire comme le vécuronium ou l'atracurium plutôt que des substances ayant des effets plus durables.
Les amino-4-quinoléines peuvent réduire l'effet de XEOMIN.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'existe pas actuellement de données appropriées sur l'utilisation de la neurotoxine botulinique de type A pendant la grossesse. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Le risque potentiel chez la femme n'est pas connu.
Par conséquent, XEOMIN ne doit pas être utilisé pendant la grossesse à moins d'une nécessité absolue et si le bénéfice potentiel justifie le risque.
Il nexiste pas de données quant à lexcrétion de la neurotoxine botulinique de type A dans le lait maternel. Par conséquent, XEOMIN ne doit pas être utilisé pendant l'allaitement.
Fertilité
Il nexiste pas de données cliniques sur lutilisation de la neurotoxine botulinique de type A. Aucun effet sur la fertilité du mâle ou de la femelle na été mis en évidence au cours des études précliniques (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Généralement, les effets indésirables se manifestent au cours de la première semaine suivant le traitement et sont de nature transitoire. Ils sont dûs à la substance active, à la technique dinjection ou aux deux.
Effets indésirables indépendants de lindication.
Effets indésirables liés à linjection
Des douleurs localisées, une inflammation, des paresthésies, une hypoesthésie, une sensibilité, un gonflement, un dème, un érythème, un prurit, une infection localisée, un hématome, un saignement et/ou des ecchymoses peuvent apparaître. La douleur liée à la piqûre et/ou à l'anxiété peut induire une réponse vagale, avec hypotension transitoire, nausée, acouphène et syncope.
Effets indésirables liés à la classe
Une faiblesse musculaire localisée est un effet pharmacologique attendu de la toxine botulinique de type A
Diffusion de la toxine
Des effets indésirables liés à une réelle diffusion de la toxine de type A à distance du site dinjection ont été très rarement rapportés (faiblesse musculaire excessive, dysphagie, pneumopathie dinhalation avec issue fatale dans certains cas) (voir section 4.4).
Réactions dhypersensibilité
Des réactions dhypersensibilité graves et/ou immédiates incluant des chocs anaphylactiques, une maladie sérique, une urticaire, un dème des tissus mous et une dyspnée ont été rarement rapportés. Certaines de ces réactions ont été décrites à la suite dinjection de toxine botulinique de type A conventionnelle soit seule soit en association avec dautres agents connus pour causer des réactions similaires.
Effets indésirables dépendants de lindication.
Torticolis spasmodique
Le traitement du torticolis spasmodique peut causer une dysphagie dintensité variable avec un risque de fausses routes nécessitant une intervention médicale. La dysphagie peut persister 2 à 3 semaines après linjection. Un cas de dysphagie persistant jusquà cinq mois a été rapporté.
Expérience clinique des effets indésirables
Sur la base des études cliniques, la fréquence des effets indésirables est donnée pour chaque indication. La fréquence est définie comme suit : très fréquent (≥1/10) ; fréquent (≥1/100 à <1/10) ; peu fréquent (≥1/1 000 à <1/100) ; rare (≥1/10 000 à <1/1 000) ; très rare (<1/10 000) ; inconnu (ne peut être estimé sur les données disponibles).
Blépharospasme
Les effets indésirables suivants ont été rapportés avec XEOMIN :
Affections du système nerveux
Fréquent : céphalées, parésie faciale
Affections oculaires
Très fréquent : ptosis, sécheresse oculaire
Fréquent : vision trouble, trouble de la vision, diplopie, larmoiement important
Affections gastro-intestinales
Fréquent : sécheresse buccale, dysphagie
Affections de la peau et du tissu sous-cutané
Peu fréquent : rash
Troubles généraux et réactions au site dadministration
Fréquent : douleur au site dinjection, fatigue
Affections musculo-squelettiques et du tissu conjonctif
Fréquent : faiblesse musculaire
Torticolis spasmodique
Les effets indésirables suivants ont été rapportés avec XEOMIN :
Affections du système nerveux
Fréquent : céphalées, présyncope, vertiges
Peu fréquent : troubles de lélocution
Affections respiratoires, thoraciques et médiastinales
Peu fréquent : dysphonie, dyspnée.
Affections gastro-intestinales
Très fréquent : dysphagie
Fréquent : sécheresse buccale, nausées
Affections de la peau et du tissu sous-cutané
Fréquent : hyperhidrose
Peu fréquent : rash
Affections musculo-squelettiques et du tissu conjonctif
Fréquent : douleur du cou, faiblesse musculaire, myalgie, spasmes musculaires, raideurs musculaires
Troubles généraux et réactions au site dadministration
Fréquent : douleur au site d'injection, asthénie
Infections et infestations
Fréquent : infection des voies aériennes supérieures
Spasticité des membres supérieurs
Les effets indésirables suivants ont été rapportés avec XEOMIN :
Affections du système nerveux
Peu fréquent : céphalées, hypoesthésie
Affections gastro-intestinales
Fréquent : bouche sèche
Peu fréquent : dysphagie, nausée
Affections musculo-squelettiques et du tissu conjonctif
Peu fréquent : faiblesse musculaire, douleur des extrémités, myalgie
Troubles généraux et réactions au site dadministration
Peu fréquent : asthénie
Inconnu : douleur au site dinjection
Certains de ces évènements indésirables peuvent être dus à la maladie.
Données après commercialisation
Des syndromes pseudo-grippaux et des réactions dhypersensibilité telles que gonflement, dème (y compris à distance du site dinjection), érythème, prurit, rash (localisé ou généralisé) et des difficultés respiratoires ont été rapportés.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Des doses importantes de toxine botulinique peuvent provoquer, par diffusion à distance du site dinjection, des paralysies neuromusculaires prononcées avec une variété de symptômes. Les symptômes peuvent se traduire par une faiblesse généralisée, un ptosis, une diplopie, des troubles de la respiration ou délocution, une paralysie des muscles respiratoires ou des troubles de la déglutition pouvant entraîner une pneumopathie d'inhalation.
Mesures en cas de surdosage
En cas de surdosage, une surveillance médicale des symptômes évocateurs de faiblesse musculaire excessive ou de paralysie musculaire doit être mise en place. Un traitement symptomatique peut être nécessaire et une assistance respiratoire peut être requise en cas de paralysie des muscles respiratoires.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : autres myorelaxants, agent à action périphérique, Code ATC : M03AX01.
La neurotoxine botulinique de type A bloque la transmission cholinergique dans la jonction neuromusculaire en inhibant la libération d'acétylcholine. Les terminaisons nerveuses de la plaque motrice ne répondent plus aux impulsions nerveuses empêchant la sécrétion des neurotransmetteurs au niveau de cette plaque motrice (dénervation chimique). La formation de nouvelles terminaisons nerveuses et de plaques motrices permet le rétablissement d'une transmission par impulsion au niveau de la plaque motrice.
Mécanisme daction
Le mécanisme d'action de la toxine botulinique de type A sur les terminaisons nerveuses cholinergiques peut être décrit par un procédé séquentiel en quatre temps faisant intervenir les étapes suivantes:
· Liaison : la chaîne lourde de la neurotoxine botulinique de type A se fixe de manière hautement sélective et avec une très forte affinité aux récepteurs cholinergiques.
· Internalisation : invagination de la membrane nerveuse terminale et encapsulation de la toxine dans la terminaison nerveuse (endocytose).
· Translocation : la fonction amine terminale de la chaîne lourde de la neurotoxine forme un pore dans la membrane de la vésicule ; le pont disulfure est rompu et la chaîne légère de la neurotoxine passe à travers ce pore dans le cytosol.
· Inhibition : lorsque la chaîne légère est libérée, elle clive de façon très sélective la protéine cible spécifique (SNAP 25) qui est indispensable à la libération d'acétylcholine.
Le rétablissement complet de la fonction motrice/conduction nerveuse intervient normalement dans les 3 à 4 mois qui suivent linjection intramusculaire, après régénération des terminaisons nerveuses et rétablissement des connexions avec la plaque motrice.
Efficacité et sécurité clinique
Léquivalence thérapeutique dune dose unique de XEOMIN a été démontrée contre le comparateur Botox (complexe donabotulinumtoxin A ou toxine botulinique de type A 900kD) dans deux études comparatives de phase 3, une réalisée chez des patients atteints de blépharospasme (étude MRZ 60201-0003, n=300) et une autre réalisée chez des patients atteints de torticolis spasmodique (étude MRZ 60201-0013, n=463). Les résultats de ces études suggèrent également que XEOMIN et ce comparateur ont des profils defficacité et de tolérance comparables chez les patients présentant un blépharospasme ou un torticolis spasmodique pour un ratio de conversion de dose de 1:1 (voir rubrique 4.2)
Blépharospasme
Une étude multicentrique de phase III, randomisée, en double-aveugle contre placebo a été réalisée chez des patients présentant un blépharospasme essentiel bénin (N=109). Les patients présentaient à linclusion un sous-score de sévérité ≥ 2 sur léchelle Jankovic (JRS) avec une réponse clinique satisfaisante et stable à un traitement par Botox(onabotulinumtoxin A) .
Les patients randomisés (2:1) étaient traités avec une dose unique de XEOMIN (n=75) ou de placebo (n=34) à la même posologie (± 10%) que les 2 sessions dinjection par Botox qui ont précédé linclusion. La dose maximale autorisée dans cette étude était de 50 unités par il avec une moyenne de 32 unités par il pour XEOMIN.
Le critère principal defficacité, défini par la modification du sous-score de sévérité sur léchelle JRS à 6 semaines par comparaison à la baseline (ITT), a été évalué en intention de traiter (ITT) sachant que les données manquantes étaient remplacées par la dernière donnée disponible du patient. La différence de -1,0 point (IC95%:-1,4;-0,5) du sous-score de sévérité sur léchelle JRS entre la baseline et à 6 semaines retrouvée entre le groupe XEOMIN et le groupe placebo, est statistiquement significative (p<0,001).
Les patients pouvaient être inclus dans la période dextension si une nouvelle injection était nécessaire. Les patients ont reçu jusquà cinq injections de XEOMIN. Un intervalle minimum dau moins 6 semaines était requis entre 2 sessions dinjection. La durée totale de létude était de 48 à 69 semaines. Au cours de cette étude, les intervalles médians des sujets traités par XEOMIN était compris entre 10,14 (1er intervalle) et 12,00 semaines (du 2nd au 5ème intervalle).
Torticolis spasmodique
Une étude multicentrique de phase III, randomisée, en double-aveugle contre placebo a été réalisée chez des patients présentant une dystonie cervicale à prédominance rotationnelle (N=233). Les patients présentaient à linclusion un score total ≥ 20 sur léchelle TWSTRS (Toronto Western Spasmodic Torticolis Rating Score). Les patients randomisés (1:1:1) étaient traités avec une dose unique de XEOMIN 240 unités (n=81), XEOMIN 120 unités (n=78) ou de placebo (n=74). Le nombre et les sites dinjection étaient déterminés par linvestigateur.
Le critère principal defficacité, défini par la modification du LS moyen à 4 semaines par comparaison à la baseline (ITT), a été évalué en intention de traiter (ITT) sachant que les données manquantes étaient remplacées par la valeur à la baseline du patient. La modification du score total TWSTRS entre la baseline et la semaine 4 était significativement plus importante dans le groupe XEOMIN que dans le groupe placebo (p<0,001 pour tous les modèles statistiques). Ces différences étaient également significatives sur le plan clinique : -9,0 points entre XEOMIN 240U et le placebo ; -7,5 points entre XEOMIN 120U et le placebo dans ce modèle statistique.
Les patients pouvaient être inclus dans la période dextension si une nouvelle injection était nécessaire. Les patients ont reçu jusquà cinq injections de XEOMIN 120U ou 240U. Un intervalle minimum dau moins 6 semaines était requis entre 2 sessions dinjection. La durée totale de létude était de 48 à 69 semaines. Au cours de cette étude, les intervalles médians des sujets traités par XEOMIN était compris entre 10,00 (1er intervalle) et 13,14 semaines (du 3ème au 6ème intervalle).
Spasticité des membres supérieurs
Dans l'étude pivot multicentrique réaliséeen double-aveugle, contre placebo chez des patients présentant une spasticité des membres supérieurs en post-AVC, 148 patients ont été randomisés : XEOMIN (n=73) ou Placebo (n=75). Les doses qui ont été administrées en traitement initial sont celles mentionnées dans le RCP (voir rubrique 4.2). La dose cumulée après traitements répétés jusqu'à 6 fois dans l'étude clinique était en moyenne de 1333 unités (maximum 2395 unités) et ce, pour une période de traitement jusquà 89 semaines.
Le critère primaire d'efficacité (taux de réponse des fléchisseurs du poignet au score d'Ashworth à la semaine 4) était défini comme une amélioration d'au moins 1 point sur l'échelle d'Ashworth en 5 points. Les patients traités par XEOMIN (taux de réponse: 68,5%) avaient 3,97 fois plus de chances d'être répondeurs en comparaison aux patients sous placebo (taux de réponse: 37,3% ; IC95% : 1,90-8,30 ; p < 0,001, population en ITT).
Cette étude à dose fixe n'a pas été conçue pour distinguer les patients de sexe féminin et masculin. Néanmoins, dans une analyse post-hoc, les taux de réponse étaient plus élevés chez la femme (89,3%) par rapport à l'homme (55,6%), la différence étant statistiquement significative pour les femmes seulement. Toutefois, les taux de réponse des hommes sur l'échelle d'Ashworth après 4 semaines de traitement par XEOMIN, étaient plus élevés dans tous les groupes musculaires traités comparativement au placebo.
Les taux de réponse étaient comparables chez l'homme et chez la femme au cours de la période de prolongation en ouvert de l'étude pivot (doses variables autorisées au cours de cette période), pour laquelle 145 patients ont été inclus et ont reçu jusqu'à 5 sessions d'injection, ainsi que dans l'étude en simple aveugle (EudraCT N°2006-003036-30) où l'efficacité et la sécurité de XEOMIN, à deux dilutions différentes, ont été évaluées chez 192 patients présentant une spasticité des membres supérieurs d'étiologies diverses.
Une autre étude clinique contrôlée de phase III, réalisée en double aveugle contre placebo, a inclus 317 patients, naïfs de tout traitement, présentant une spasticité du membre supérieur post AVC depuis au moins 3 mois. Au cours de la phase principale, une dose totale de 400 unités de XEOMIN a été injectée par voie intramusculaire en fonction de lobjectif clinique principal choisi entre la flexion du coude, la flexion du poignet ou le poing serré, ainsi que dans tout autre groupe musculaire atteint (n=210). Lanalyse du critère principal et de lensemble des critères defficacité à 4 semaines ont démontré une amélioration significative du taux de répondeurs (score dAshworth), du score dAshworth versus baseline et de limpression globale de linvestigateur.
296 patients ayant finalisé la phase principale ont été inclus dans la phase dextension en ouvert (OLEX) et ont reçu jusquà 3 injections à chaque cycle. Les patients étaient traités avec une seule dose totale de 400 unités de XEOMIN répartie à lensemble des muscles affectés puis suivis 12 semaines. La durée totale de létude était de 48 semaines.
Une étude de phase III en ouvert, évaluant, entre autres, le traitement des muscles de lépaule, a inclus 155 patients présentant une spasticité des membres supérieurs et des membres inférieurs. Des doses jusquà 600 unités de XEOMIN dans le membre supérieur pouvaient être administrées. Cette étude a montré une corrélation positive entre laugmentation des doses de XEOMIN et lamélioration des patients ; amélioration objectivée par le score dAshworth et par dautres variables defficacité ; sans modification de la sécurité des patients ou du profil de tolérance de XEOMIN.
Population pédiatrique
Lagence européenne des médicaments a accordé une dérogation à lobligation de soumettre les résultats détudes réalisées avec XEOMIN dans tous les sous-groupes de la population pédiatrique dans le traitement de la dystonie et chez le nouveau-né et les enfants entre 0 et 24 mois dans le traitement de la spasticité (voir rubrique 4.2 pour les informations concernant lusage pédiatrique).
5.2. Propriétés pharmacocinétiques
Caractéristiques générales de la substance active
Les études de distribution et de cinétique classiques ne peuvent pas être effectuées avec la neurotoxine botulinique de type A car la substance active est administrée en très petites quantités (picogrammes par injection) et se lie rapidement et de manière irréversible aux terminaisons nerveuses cholinergiques.
La toxine botulinique naturelle de type A est un complexe de haut poids moléculaire qui, en plus de la neurotoxine (150 kD), contient d'autres protéines non toxiques hémagglutinantes et non hémagglutinantes. Contrairement à la toxine botulinique de type A conventionnelle, XEOMIN contient la neurotoxine pure (150 kD) puisque celle-ci est dénuée de protéines complexantes et de ce fait, contient peu de protéines étrangères. La teneur en protéines étrangères administrée est considérée comme lun des facteurs déchec secondaire au traitement.
La neurotoxine botulinique de type A a montré qu'elle subissait un transport axonal rétrograde après injection intramusculaire. Le passage transsynaptique rétrograde de la neurotoxine botulinique active de type A dans le système nerveux central n'a cependant pas été observé aux doses thérapeutiques préconisées.
La neurotoxine botulinique de type A liée aux récepteurs membranaires est endocytosée dans la terminaison nerveuse avant d'atteindre sa cible (SNAP 25) puis est dégradée par voie intracellulaire. Les molécules de la neurotoxine botulinique de type A circulant librement, qui n'ont pas été liées aux récepteurs des terminaisons nerveuses cholinergiques présynaptiques, subissent un processus de phagocytose ou de pinocytose et sont dégradées comme toute autre protéine circulant librement.
Distribution
En raison de la nature du produit, aucune étude cinétique n'a été conduite chez l'homme.
5.3. Données de sécurité préclinique
Les conclusions des études de toxicité à doses répétées chez lanimal telles quatonie, parésie et atrophie du muscle injecté, étaient essentiellement liées à l'activité pharmacodynamique.
Aucun signe d'intolérance locale n'a été noté. Les études de toxicité sur la reproduction réalisées avec XEOMIN chez le lapin nont pas montré de toxicité sur la fertilité du mâle ou de la femelle ni deffet direct sur le développement embryo-ftal ou sur le développement pre- et post-natal chez le rat et/ou le lapin. Cependant des études dembryotoxicité, après administration quotidienne, hebdomadaire ou bihebdomadaire de XEOMIN à une femelle, à des doses exposant la femelle à une perte de poids ; une augmentation du nombre d'avortements chez le lapin et une légère diminution du poids du ftus chez le rat ont été observées. Une exposition systémique en continu de femelles gravides pendant la phase sensible (inconnue) de l'organogenèse comme pré-requis pour l'induction d'effets tératogènes ne peut pas nécessairement être envisagée dans ces études.
En conséquence, les marges de sécurité cliniques étaient généralement étroites à fortes doses.
Aucune étude de génotoxicité ou de carcinogénicité n'a été conduite avec XEOMIN.
Saccharose.
Flacon fermé: 3 ans
Solution reconstituée : la stabilité physique et chimique en cours dutilisation a été démontrée pendant 24 heures entre 2°C et 8°C. D'un point de vue microbiologique, le produit doit être utilisé immédiatement. Dans le cas contraire, la durée et les conditions de conservation de la solution reconstituée relèvent de la responsabilité de lutilisateur et ne devraient pas excéder 24 heures à une température comprise entre +2°C et +8°C, à moins que la reconstitution nait été effectuée dans des conditions dasepsie maitrisées et validées.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
Pour les conditions de conservation après reconstitution du médicament, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon (verre de type 1), muni dun bouchon (caoutchouc bromobutyle) et dune bague de sécurité (aluminium).
Boîtes de 1, 2, 3 ou 6 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Reconstitution
XEOMIN est reconstitué avant utilisation avec une solution injectable de chlorure de sodium à 9 mg/ml (0,9 %). La reconstitution et la dilution doivent être réalisées selon les bonnes pratiques cliniques, particulièrement dans le respect de l'asepsie.
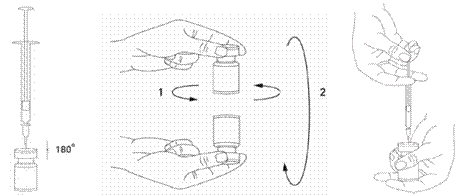
Il est de bonne pratique de reconstituer le produit et préparer les seringues sur des supports papier doublés de plastique pour recevoir tout déversement accidentel. Un volume approprié de solution de chlorure de sodium est aspiré dans une seringue (voir tableau de dilution). Une aiguille courte en biseau de 20-27 Gauge est recommandée pour la reconstitution. Après insertion verticale de laiguille à travers le bouchon en caoutchouc, le solvant est injecté délicatement dans le flacon afin déviter la formation de mousse. Le flacon doit être jeté si la dépression n'entraîne pas l'aspiration du solvant à l'intérieur du flacon. Retirer la seringue du flacon et mélanger délicatement XEOMIN au solvant par des mouvements circulaires et par retournement du flacon. Ne pas mélanger vigoureusement. Si besoin, laiguille ayant servi à la reconstitution de la solution peut rester dans le flacon. Le volume de produit nécessaire est aspiré avec une nouvelle seringue stérile adaptée à linjection.
|
|
La solution reconstituée de XEOMIN est limpide et incolore.
XEOMIN ne doit pas être utilisé si la solution reconstituée présente un aspect trouble ou si elle contient des particules ou des matières floconneuses.
Afin de prévenir tout surdosage accidentel, il convient de bien sassurer lors de la reconstitution du produit que le volume de solvant est adapté à la présentation choisie. Si différentes présentations sont utilisées au cours dune même séance dinjection, il convient de sassurer que le volume de solvant utilisé lors de la reconstitution du produit donne le bon nombre dunités par 0,1ml. Le volume de solvant à ajouter diffère entre XEOMIN 50 unités, XEOMIN 100 unités et XEOMIN 200 unités. Chaque seringue utilisée doit être identifiée en conséquence.
Les dilutions possibles pour XEOMIN 50 sont indiquées dans le tableau suivant :
|
Dose obtenue (unités par 0,1 ml) |
Solvant ajouté (solution injectable de chlorure de sodium à 9 mg/ml (0,9%)) |
|
|
· 20 unités |
· 0,25ml |
|
|
· 10 unités |
· 0,5ml |
|
|
· 8 unités |
· 0,625 ml |
|
|
· 5 unités |
· 1ml |
|
|
· 4 unités |
· 1,25 ml |
|
|
· 2,5 unités |
· 2ml |
|
|
· 2 unités |
· 2,5 ml |
|
|
· 1,25 unités |
· 4ml |
|
Toute solution injectable qui a été conservée pendant plus de 24 heures et toute fraction de solution injectable restante ou non utilisée doivent être jetées.
Procédure à suivre pour une élimination en toute sécurité des flacons, des seringues et matériels utilisés.
Tous flacons non utilisés ou contenant des solutions reconstituées résiduelles et/ou les seringues peuvent être autoclavés.
Il est également possible dinactiver toute fraction restante de XEOMIN par addition déthanol à 70%, disopropanol à 50%, de lauryl sulfate de sodium à 0,1% (détergent anionique), dune solution diluée d'hydroxyde de sodium (NaOH 0,1 N) ou dune solution diluée d'hypochlorite de sodium (NaOCl à au moins 0,1%).
Les flacons utilisés, seringues et matériels ne doivent pas être vidés mais doivent être jetés dans des récipients adaptés et éliminés conformément à la règlementation locale en vigueur.
Recommandations en cas d'incident lors de la manipulation de la toxine botulinique de type A
· Toute projection doit être essuyée avec un matériel absorbant imbibé d'une des solutions listées ci-dessus pour la poudre ou bien avec un matériel absorbant sec pour le produit reconstitué.
· Les surfaces contaminées sont nettoyées avec un matériel absorbant, imbibé d'une des solutions listées ci-dessus puis séchées.
· Si le flacon est brisé, procéder comme indiqué ci-dessus au ramassage méticuleux des particules de verre et essuyer le produit, en évitant toute coupure cutanée.
· En cas de contact avec la peau, rincer abondamment la zone touchée à l'eau.
· En cas de contact avec les yeux, rincer abondamment avec de l'eau ou avec une solution pour lavage ophtalmique.
· En cas de contact du produit avec une blessure, une coupure ou une piqûre, rincer abondamment avec de l'eau et prendre les mesures médicales appropriées en fonction de la dose injectée.
Ces instructions d'utilisation, de manipulation et d'élimination doivent être scrupuleusement suivies.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
ECKENHEIMER LANDSTRASSE 100
60318 FRANKFURT / MAIN Allemagne
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
34009 580 884 7 6 : 50 unités de neurotoxine de Clostridium botulinum de type A en flacon (verre de type I), muni d'un bouchon (caoutchouc bromobutyle) et d'une bague de sécurité (aluminium), boîte de 1 flacon.
34009 580 885 3 7 : 50 unités de neurotoxine de Clostridium botulinum de type A en flacon (verre de type I), muni d'un bouchon (caoutchouc bromobutyle) et d'une bague de sécurité (aluminium), boîte de 2 flacons.
34009 580 887 6 6 : 50 unités de neurotoxine de Clostridium botulinum de type A en flacon (verre de type I), muni d'un bouchon (caoutchouc bromobutyle) et d'une bague de sécurité (aluminium), boîte de 3 flacons.
34009 580 888 2 7 : 50 unités de neurotoxine de Clostridium botulinum de type A en flacon (verre de type I), muni d'un bouchon (caoutchouc bromobutyle) et d'une bague de sécurité (aluminium), boîte de 6 flacons.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I. Médicament réservé à l'usage hospitalier.