
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 24/01/2018
ERLOTINIB TEVA 100 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Erlotinib (sous forme de chlorhydrate derlotinib).................................................................... 100 mg
Pour un comprimé pelliculé.
Excipient à effet notoire : chaque comprimé pelliculé contient 110,125 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé blanc, rond, biconvexe, comportant la mention « 100 » sur une face, et mesurant environ 10,1 x 4,1 mm.
4.1. Indications thérapeutiques
Cancer Bronchique Non à Petites Cellules (CBNPC) :
ERLOTINIB TEVA est indiqué en première ligne de traitement des formes localement avancées ou métastatiques du cancer bronchique non à petites cellules (CBNPC) chez les patients présentant des mutations activatrices de lEGFR.
ERLOTINIB TEVA est également indiqué dans le traitement de switch maintenance des formes localement avancées ou métastatiques du CBNPC chez les patients avec mutation activatrice de lEGFR et présentant une maladie stable après une première ligne de chimiothérapie.
ERLOTINIB TEVA est également indiqué dans le traitement des formes localement avancées ou métastatiques du CBNPC après échec d'au moins une ligne de chimiothérapie.
Lors de la prescription dERLOTINIB TEVA, les facteurs associés à une survie prolongée doivent être pris en considération.
Aucun bénéfice en survie ou autres effets cliniquement significatifs du traitement nont été démontrés chez les patients dont lexpression du récepteur au facteur de croissance épidermique (EGFR) de la tumeur (déterminée par IHC) était négative (voir rubrique 5.1).
Cancer du pancréas :
ERLOTINIB TEVA, en association à la gemcitabine, est indiqué dans le traitement du cancer du pancréas métastatique.
Lors de la prescription dERLOTINIB TEVA, les facteurs associés à une survie prolongée doivent être pris en considération (voir rubriques 4.2 et 5.1).
Aucun avantage en survie na été montré chez les patients ayant une maladie localement avancée.
4.2. Posologie et mode d'administration
Posologie
Le traitement par ERLOTINIB TEVA doit être supervisé par un médecin expérimenté dans l'utilisation des traitements anticancéreux.
Patients atteints dun Cancer Bronchique Non à Petites Cellules :
La recherche de mutation de lEGFR doit être effectuée avant linitiation du traitement par ERLOTINIB TEVA chez les patients atteints de CBNPC localement avancé ou métastatique nayant pas préalablement reçu de traitement par chimiothérapie.
La posologie quotidienne recommandée dERLOTINIB TEVA est de 150 mg à prendre au moins une heure avant ou deux heures après un repas.
Patients atteints dun cancer du pancréas :
La posologie quotidienne recommandée dERLOTINIB TEVA est de 100 mg à prendre au moins une heure avant ou deux heures après un repas, en association à la gemcitabine (voir le résumé des caractéristiques de la gemcitabine dans le cancer du pancréas). Chez les patients qui ne développent pas déruptions cutanées dans les 4 à 8 premières semaines de traitement, la poursuite du traitement par ERLOTINIB TEVA doit être réévaluée (voir rubrique 5.1).
Quand une adaptation de la posologie est nécessaire, la dose doit être réduite par paliers de 50 mg (voir rubrique 4.4).
ERLOTINIB TEVA est disponible en dosages de 25 mg, 100 mg et 150 mg.
Ladministration conjointe de substrats et de modulateurs du CYP3A4 peut nécessiter une adaptation de la dose (voir rubrique 4.5).
Insuffisants hépatiques : Lerlotinib est éliminé par métabolisme hépatique et excrétion biliaire. Bien que lexposition à lerlotinib était similaire chez les patients ayant une insuffisance hépatique modérée (score de Child-Pugh 7-9) par rapport aux patients ayant une fonction hépatique adéquate, ERLOTINIB TEVA devra être utilisé avec précaution chez les patients présentant une insuffisance hépatique. Une réduction de la posologie ou une interruption dERLOTINIB TEVA devrait être envisagée en cas de survenue deffets indésirables graves. La tolérance et lefficacité de lerlotinib nont pas été étudiées chez les patients présentant un trouble hépatique sévère (ASAT/SGOT et ALAT/SGPT > 5 fois la limite supérieure de la normale). Lutilisation dERLOTINIB TEVA chez les patients ayant un trouble hépatique sévère nest pas recommandée (voir rubrique 5.2).
Insuffisants rénaux : La sécurité et lefficacité de lerlotinib nont pas été étudiées chez les patients insuffisants rénaux (créatinémie >1,5 fois la limite supérieure de la normale). Sur la base des données de pharmacocinétique, aucune adaptation de la posologie ne semble nécessaire chez les patients ayant une insuffisance rénale légère ou modérée (voir rubrique 5.2). Lutilisation dERLOTINIB TEVA nest pas recommandée chez les patients ayant une insuffisance rénale sévère.
Population pédiatrique : La sécurité et lefficacité de lerlotinib chez des patients âgés de moins de 18 ans nont pas été établies. Lutilisation dERLOTINIB TEVA en pédiatrie nest pas recommandée.
Fumeurs : Il a été montré que le tabagisme réduit lexposition à lerlotinib de 50-60 %. La dose maximale tolérée dERLOTINIB TEVA chez les patients ayant un CBNPC et qui fument des cigarettes est de 300 mg. Lefficacité et la tolérance à long terme dune posologie plus élevée que la posologie initiale recommandée nont pas été établis chez les patients qui continuent à fumer des cigarettes (voir rubriques 4.5 et 5.2). Par conséquent, les fumeurs devront être encouragés à arrêter de fumer, compte tenu de la réduction des concentrations plasmatiques derlotinib chez les fumeurs par rapport aux non-fumeurs.
Hypersensibilité à lerlotinib ou à lun des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Evaluation du statut de la mutation de lEGFR
Lors de lévaluation du statut de la mutation de lEGFR dun patient, il est important de choisir une méthode robuste et bien validée pour éviter les faux négatifs et les faux positifs.
Fumeurs
Les fumeurs devront être encouragés à arrêter de fumer, compte tenu de la réduction des concentrations plasmatiques derlotinib chez les fumeurs par rapport aux non-fumeurs. Le degré de réduction est probablement cliniquement significatif (voir rubrique 4.5).
Affections pulmonaires interstitielles
Peu fréquemment, des évènements à type daffections pulmonaires interstitielles (API), dont certains fatals, ont été décrits chez des patients traités par erlotinib pour un cancer bronchique non à petites cellules (CBNPC), un cancer du pancréas ou dautres tumeurs solides à un stade avancé. Au cours de létude pivot BR.21 dans le CBNPC, lincidence des cas dAPI (0,8 %) a été identique dans les groupes erlotinib et placebo. Lors de létude menée dans le cancer du pancréas en association à la gemcitabine, lincidence des événements à type dAPI était de 2,5 % dans le groupe erlotinib plus gemcitabine contre 0,4 % dans le groupe gemcitabine plus placebo. Lincidence globale chez lensemble des patients traités par erlotinib au cours des études (dont des études non contrôlées et des études avec chimiothérapie concomitante) a été denviron 0,6 % comparée à 0,2 % chez les patients recevant un placebo. Chez les patients avec suspicion dévénements à type dAPI, les diagnostics reportés incluaient notamment : pneumopathie inflammatoire, pneumopathie radique, pneumopathie dhypersensibilité, pneumonie interstitielle, affection pulmonaire interstitielle, bronchiolite obstructive, fibrose pulmonaire, Syndrome de Détresse Respiratoire Aiguë (SDRA), alvéolite inflammatoire et infiltration pulmonaire. Les symptômes sont survenus quelques jours voire plusieurs mois après linstauration du traitement par erlotinib. La plupart des cas ont été associés à des facteurs confondants ou favorisants tels qu'une chimiothérapie concomitante ou antérieure, une radiothérapie antérieure, une atteinte préexistante du parenchyme pulmonaire, des métastases pulmonaires ou des infections respiratoires. Une incidence plus élevée dAPI (environ 5 % avec un taux de mortalité de 1,5 %) est observée chez les patients dorigine japonaise.
Chez les patients qui présentent de manière inexpliquée de nouveaux symptômes pulmonaires et/ou une majoration de ces symptômes tels que dyspnée, toux et fièvre, le traitement par erlotinib doit être interrompu dans l'attente dexplorations diagnostiques. Les patients traités par erlotinib associé à la gemcitabine doivent être étroitement surveillés quant à la possibilité de développer un évènement à type dAPI. En cas de diagnostic dAPI, le traitement par ERLOTINIB TEVA doit être arrêté et un traitement adéquat doit être instauré si nécessaire (voir rubrique 4.8).
Diarrhées, déshydratation, déséquilibre des électrolytes et insuffisance rénale
Des cas de diarrhée (dont de très rares cas fatals) sont survenus chez environ 50 % des patients traités par erlotinib ; les formes modérées ou sévères doivent être traitées, par exemple, par le lopéramide. Une réduction de la posologie peut parfois être nécessaire. Dans les études cliniques, les doses étaient réduites par paliers de 50 mg. Les réductions de doses par paliers de 25 mg nont pas été étudiées. En cas de déshydratation associée à des diarrhées, à des nausées, à une anorexie ou à des vomissements sévères et persistants, le traitement par ERLOTINIB TEVA doit être interrompu et des mesures adaptées de réhydratation doivent être instaurées (voir rubrique 4.8). De rares cas dhypokaliémie et dinsuffisance rénale (dont certains dévolution fatale) ont été rapportés. Certains cas étaient secondaires à une déshydratation sévère due à des diarrhées, des vomissements et/ou une anorexie, alors que dautres cas étaient liés à une chimiothérapie concomitante. Dans les cas de diarrhées sévères ou persistantes, ou conduisant à une déshydratation, en particulier chez les patients ayant des facteurs de risques aggravants (en particulier en cas de chimiothérapie concomitante et dautres traitements, symptômes ou pathologies ou autres facteurs prédisposants dont lâge), le traitement par ERLOTINIB TEVA doit être interrompu et des mesures appropriées de réhydratation intensive du patient par voie intraveineuse doivent être mises en uvre. De plus la fonction rénale et les électrolytes sériques, incluant la kaliémie, doivent être surveillés chez les patients à risque de déshydratation.
Hépatite, insuffisance hépatique
De rares cas dinsuffisance hépatique (dont certains dévolution fatale) ont été rapportés au cours du traitement par erlotinib. Des facteurs tels que des antécédents de troubles hépatiques ou des traitements hépatotoxiques concomitants ont été associés. Par conséquent, chez ces patients, des tests réguliers de la fonction hépatique doivent être envisagés. Ladministration derlotinib doit être interrompue en cas de modifications importantes de la fonction hépatique (voir rubrique 4.8). Lerlotinib nest pas recommandé chez les patients ayant un trouble hépatique sévère.
Perforation gastro-intestinale
Les patients recevant lerlotinib ont un risque augmenté de perforation gastro-intestinale, qui a été peu fréquemment observée (dont certains cas ont été fatals). Les patients recevant de façon concomitante des agents anti-angiogéniques, des corticostéroïdes, des AINS, et/ou une chimiothérapie à base de taxane, ou un antécédent dulcère gastro-duodénal ou de diverticulose ont un risque augmenté. Lerlotinib doit être arrêté définitivement chez les patients qui développent une perforation gastro-intestinale (voir rubrique 4.8).
Lésions bulleuses et exfoliatives
Des cas de lésions bulleuses, phlycténulaires et exfoliatives ont été rapportés, y compris de très rares cas suggérant un syndrome de Stevens-Johnson / Syndrome de Lyell (nécrolyse épidermique toxique), qui dans certains cas ont été fatals (voir rubrique 4.8). Le traitement par erlotinib doit être interrompu ou arrêté définitivement si les patients présentent des lésions bulleuses ou exfoliatives sévères. Les patients présentant des lésions bulleuses et exfoliatives doivent être explorés à la recherche dune infection cutanée et traités selon les recommandations locales.
Affections oculaires
Les patients présentant des signes et des symptômes évocateurs d'une kératite aiguë ou dune kératite saggravant tels que, inflammation oculaire, larmoiement, sensibilité à la lumière, vision floue, douleur oculaire et/ou yeux rouges, doivent être adressés rapidement à un spécialiste en ophtalmologie. Si un diagnostic de kératite ulcérée est confirmé, le traitement par erlotinib doit être interrompu ou arrêté. Si une kératite est diagnostiquée, les bénéfices et les risques de la poursuite du traitement devront être soigneusement évalués. Lerlotinib doit être utilisé avec prudence chez les patients ayant des antécédents de kératite, kératite ulcérée ou sécheresse oculaire sévère. L'utilisation de lentilles de contact est également un facteur de risque de kératite et dulcération. De très rares cas de perforation ou dulcération de la cornée ont été rapportés lors de lutilisation derlotinib (voir rubrique 4.8).
Interactions avec dautres médicaments
Les inducteurs puissants du CYP3A4 peuvent réduire lefficacité de lerlotinib tandis que les inhibiteurs puissants du CYP3A4 peuvent augmenter sa toxicité. La prise concomitante de ce type de molécules doit être évitée (voir rubrique 4.5).
Autres interactions
Lerlotinib se caractérise par une diminution de solubilité à un pH supérieur à 5. Les médicaments qui modifient le pH de la partie supérieure du tractus gastro-intestinal, comme les inhibiteurs de la pompe à protons, les antagonistes H2 et les antiacides, peuvent modifier la solubilité de lerlotinib et de ce fait sa biodisponibilité. Laugmentation de la posologie derlotinib lors de sa co-administration à de tels produits ne compense probablement pas la diminution de son exposition. Lassociation de lerlotinib aux inhibiteurs de la pompe à protons doit être évitée. Les effets de ladministration concomitante de lerlotinib à des antagonistes H2 et à des antiacides ne sont pas connus ; cependant, une diminution de la biodisponibilité est probable. Par conséquent, ladministration concomitante de ces associations doit être évitée (voir rubrique 4.5). Si lutilisation des antiacides est jugée nécessaire durant le traitement par erlotinib, ils doivent être pris au moins 4 heures avant ou 2 heures après la dose quotidienne derlotinib.
Ce médicament contient du lactose. Son utilisation est déconseillée chez les patients présentant une intolérance au galactose, un déficit en lactase de Lapp ou un syndrome de malabsorption du glucose ou du galactose (maladies héréditaires rares).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les études dinteraction nont été réalisées que chez ladulte.
Lerlotinib et les autres substrats de CYP
In vitro l'erlotinib est un inhibiteur puissant du CYP1A1 et un inhibiteur modéré des CYP3A4 et CYP2C8, ainsi quun inhibiteur puissant de la glucuroconjugaison par lUGT1A.
Du fait de la très faible expression du CYP1A1 dans les tissus humains, la pertinence physiologique dune forte inhibition du CYP1A1 nest pas connue.
Lors de la co-administration de lerlotinib à la ciprofloxacine, un inhibiteur modéré du CYP1A2, laire sous la courbe (ASC) de lerlotinib a augmentée significativement de 39 % tandis quaucun changement significatif de la Cmax na été trouvé. De la même manière, lASC et la Cmax du métabolite actif étaient respectivement augmentées denviron 60 % et 48 %. La pertinence clinique de cette augmentation na pas été établie. Une attention particulière doit être exercée lors de lassociation de la ciprofloxacine ou des inhibiteurs puissants du CYP1A2 à lerlotinib (ex : fluvoxamine). Si des effets indésirables liés à lerlotinib sont observés, la posologie derlotinib peut être diminuée.
Le prétraitement ou la co-administration dERLOTINIB TEVA nont pas modifié la clairance des substrats spécifiques du CYP3A4, tel que le midazolam et lérythromycine, mais semblent diminuer la biodisponibilité orale du midazolam jusquà 24 %. Dans une autre étude clinique, l'erlotinib na pas modifié les paramètres pharmacocinétiques du paclitaxel, un substrat des CYP3A4/2C8, administré concomitamment. Des interactions significatives avec la clairance dautres substrats du CYP3A4 sont par conséquent improbables.
Linhibition de la glucuroconjugaison pourrait entraîner des interactions avec les médicaments substrats de lUGT1A1 et qui sont exclusivement éliminés par cette voie. Les patients avec une faible expression de lUGT1A1 ou qui présentent des troubles génétiques de la glucuroconjugaison (ex : maladie de Gilbert) pourraient présenter une augmentation des concentrations sériques en bilirubine et devront être traités avec précaution.
Chez l'Homme, lerlotinib est métabolisé par les cytochromes hépatiques, principalement par le CYP3A4 et à un moindre degré par le CYP1A2. Le métabolisme extra hépatique par le CYP3A4 intestinal, le CYP1A1 pulmonaire et le CYP1B1 du tissu tumoral contribuent potentiellement à la clairance métabolique de lerlotinib. Des interactions pourraient survenir avec les principes actifs métabolisés par ces enzymes, ou qui les inhibent ou les induisent.
Les inhibiteurs puissants du CYP3A4 ralentissent le métabolisme de lerlotinib et augmentent ses concentrations plasmatiques. Dans une étude clinique, lutilisation concomitante du kétoconazole (200 mg par voie orale deux fois par jour pendant 5 jours), un inhibiteur puissant du CYP3A4, a entraîné une augmentation de 86 % de laire sous la courbe [ASC] et de 69 % de la Cmax de lerlotinib. De ce fait, lassociation derlotinib aux inhibiteurs puissants du CYP3A4, tels que les antifongiques azolés (ex : kétoconazole, itraconazole, voriconazole), les inhibiteurs de protéase, lérythromycine ou la clarithromycine doit être faite avec prudence. Si nécessaire, la dose derlotinib doit être réduite, particulièrement en cas dapparition de toxicité.
Les inducteurs puissants du CYP3A4 accélèrent le métabolisme de lerlotinib et diminuent significativement ses concentrations plasmatiques. Dans une étude clinique, lutilisation concomitante derlotinib et de rifampicine (600 mg par voie orale une fois par jour pendant 7 jours), inducteur puissant du CYP3A4, a conduit à une diminution de 69 % de la médiane de lASC de lerlotinib. La co-administration de la rifampicine à une dose unique de 450 mg derlotinib a conduit à une moyenne de lASC de lerlotinib correspondant à 57,5 % de celle obtenue avec une dose unique de 150 mg derlotinib en labsence de rifampicine. Par conséquent, la co-administration derlotinib à des inducteurs du CYP3A4 doit être évitée. Pour les patients nécessitant un traitement concomitant derlotinib avec un puissant inducteur du CYP3A4 comme la rifampicine, une augmentation de la dose jusquà 300 mg doit être envisagée tout en surveillant étroitement leur tolérance (notamment surveillance des fonctions rénales, hépatiques et des électrolytes sériques). Si cette dose est bien tolérée pendant plus de 2 semaines, une augmentation supplémentaire jusquà la dose de 450 mg pourrait être envisagée avec une surveillance étroite de la tolérance. La diminution de lexposition à lerlotinib pourrait également apparaître avec dautres inducteurs tels que la phénytoïne, la carbamazépine, les barbituriques ou le millepertuis (hypericum perforatum). La prudence est de rigueur lorsque ces principes actifs sont associés à lerlotinib. Des traitements alternatifs faiblement inducteurs du CYP3A4 doivent être envisagés chaque fois que possible.
Lerlotinib et les anticoagulants coumariniques
Des interactions avec des dérivés coumariniques, notamment la warfarine, ayant conduit à une augmentation de lINR (International Normalized Ratio) et à des hémorragies, dans certains cas fatales, ont été rapportées chez des patients recevant lerlotinib. Chez les patients conjointement traités par un dérivé coumarinique, le temps de prothrombine ou lINR doivent être régulièrement contrôlés.
Lerlotinib et les statines
Lassociation derlotinib avec une statine peut augmenter le risque de myopathie induite par les statines (y compris rhabdomyolyse), qui a été rarement observée.
Lerlotinib et les fumeurs
Les résultats dune étude dinteraction pharmacocinétique ont montré une diminution significative de laire sous la courbe (ASCinf), de la concentration plasmatique maximale (Cmax) et de la concentration plasmatique à 24 heures respectivement dun facteur de 2,8, 1,5, et 9 après ladministration derlotinib chez les fumeurs par rapport aux non-fumeurs (voir rubrique 5.2). Par conséquent, les patients continuant à fumer devront être encouragés à arrêter le plus tôt possible avant le début du traitement par erlotinib, compte-tenu de la réduction des concentrations plasmatiques derlotinib. Leffet clinique de cette diminution dexposition na pas été évalué de façon formelle mais est probablement cliniquement significatif.
Lerlotinib et les inhibiteurs de la glycoprotéine-P
Lerlotinib est un substrat de la glycoprotéine-P. Ladministration concomitante des inhibiteurs de la glycoprotéine-P tels que la ciclosporine et le vérapamil, peut conduire à une altération de la distribution et/ou de lélimination de lerlotinib. Les conséquences de cette interaction, par exemple au niveau de la toxicité pour le SNC, nont pas été établies. Une attention particulière doit être exercée dans de telles situations.
Lerlotinib et les médicaments qui modifient le pH
Lerlotinib se caractérise par une diminution de solubilité à un pH supérieur à 5. Les médicaments qui modifient le pH de la partie supérieure du tractus gastro-intestinal peuvent modifier la solubilité de lerlotinib et de ce fait sa biodisponibilité. La co-administration de lerlotinib à loméprazole, un inhibiteur de la pompe à protons (IPP), a diminué laire sous la courbe (ASC) et la concentration maximale (Cmax) de lerlotinib respectivement de 46 % et 61 %. Il ny avait pas de modification du Tmax ou de la demi-vie. Ladministration concomitante derlotinib à 300 mg de ranitidine, un antagoniste du récepteur H2, a diminué laire sous la courbe (ASC) et la concentration maximale (Cmax) de lerlotinib respectivement de 33 % et 54 %. Laugmentation de la posologie derlotinib lors de sa co-administration à de tels produits, ne compense probablement pas la diminution de son exposition. Cependant, lorsque lerlotinib administré de façon espacée, 2 heures avant ou 10 heures après ladministration de ranitidine 150 mg deux fois par jour a été dosé, laire sous la courbe (ASC) et la concentration maximale (Cmax) de lerlotinib ont seulement diminué respectivement de 15 % et 17 %. Leffet des antiacides sur labsorption de lerlotinib na pas été étudié, mais labsorption peut être altérée, conduisant à une diminution des taux plasmatiques. En résumé, lassociation de lerlotinib aux inhibiteurs de la pompe à protons doit être évitée. Si lutilisation des antiacides est jugée nécessaire durant le traitement par erlotinib, ils doivent être pris au moins 4 heures avant ou 2 heures après la dose quotidienne derlotinib. Si lutilisation de la ranitidine est envisagée, elle doit lêtre de façon espacée ; par ex. lerlotinib doit être pris au moins 2 heures avant ou 10 heures après la ranitidine.
Lerlotinib et la gemcitabine
Dans une étude de phase Ib, il ny a eu aucun effet significatif de la gemcitabine sur les paramètres pharmacocinétiques de lerlotinib ni de lerlotinib sur ceux de la gemcitabine.
Lerlotinib et le carboplatine/paclitaxel
Lerlotinib augmente les concentrations en sel de platine. Dans une étude clinique, lutilisation concomitante de lerlotinib au carboplatine et au paclitaxel a conduit à une augmentation de 10,6 % de lASC0-48 du sel de platine total. Bien que statistiquement significative, limportance de cette différence nest pas considérée comme cliniquement pertinente. En pratique clinique, dautres facteurs associés peuvent conduire à une augmentation de lexposition au carboplatine comme une altération de la fonction rénale. Il ny a pas eu deffets significatifs du carboplatine ou du paclitaxel sur les paramètres pharmacocinétiques de lerlotinib.
Lerlotinib et la capécitabine
La capécitabine peut augmenter les concentrations de lerlotinib. Lorsque lerlotinib a été associé à la capécitabine, il y a eu une augmentation significative de lASC de lerlotinib et une augmentation limitée de la Cmax par rapport aux valeurs observées dans une autre étude dans laquelle lerlotinib a été administré seul. Il ny a pas eu deffets significatifs de lerlotinib sur les paramètres pharmacocinétiques de la capécitabine.
L'erlotinib et les inhibiteurs du protéasome
Compte tenu de leur mécanisme daction, les inhibiteurs du protéasome, y compris le bortézomib, pourraient avoir une influence sur l'effet des inhibiteurs de l'EGFR, notamment l'erlotinib. Cette influence est étayée par des données cliniques limitées et des études précliniques montrant une dégradation de lEGFR par le protéasome.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il nexiste pas de données suffisantes relatives à lutilisation derlotinib chez la femme enceinte. Les études réalisées chez lanimal nont pas mis en évidence de tératogénicité ou de parturition anormale. Cependant, un effet indésirable sur la grossesse ne peut être exclu car des études réalisées chez le rat et le lapin ont montré une létalité embryo-ftale augmentée (voir rubrique 5.3). Le risque potentiel chez l'homme est inconnu.
Femmes en âge de procréer
Les femmes en âge de procréer doivent être incitées à éviter une grossesse pendant le traitement par erlotinib. Une méthode de contraception efficace doit être utilisée pendant le traitement et pendant au moins les 2 semaines qui suivent la fin de celui-ci. En cas de survenue dune grossesse, le traitement ne doit être poursuivi que si le bénéfice attendu pour la mère justifie le risque pris pour le ftus.
En labsence de données sur lexcrétion de lerlotinib dans le lait maternel et en raison des dangers potentiels pour le nourrisson, lallaitement est déconseillé lors dun traitement par erlotinib.
Fertilité
Les études réalisées chez lanimal nont pas mis en évidence de trouble de la fertilité. Cependant, un effet indésirable sur la fertilité ne peut être exclu car les études réalisées chez lanimal ont montré des effets sur les paramètres de la reproduction (voir rubrique 5.3). Le risque potentiel chez l'Homme est inconnu.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Cancer bronchique non à petites cellules (erlotinib en monothérapie) :
Dans une étude randomisée en double aveugle (BR.21 : erlotinib en deuxième ligne de traitement), les effets indésirables (EI) les plus fréquemment observés ont été des éruptions cutanées (75 %) et des diarrhées (54 %). La plupart ont été de grade 1/2 et nont pas nécessité dintervention spécifique. Des éruptions cutanées et des diarrhées de grade 3/4 sont survenues chez respectivement 9 % et 6 % des patients traités par lerlotinib et ont conduit à des sorties détude chez 1 % des patients. Une réduction de la posologie a été nécessaire en raison dune éruption cutanée ou dune diarrhée chez respectivement 6 % et 1 % des patients. Dans létude BR.21, le délai moyen de survenue des éruptions cutanées a été de 8 jours et celui des diarrhées de 12 jours.
De manière générale, léruption cutanée se manifeste comme un érythème léger à modéré et une éruption papulopustuleuse, qui peut survenir ou saggraver au niveau des zones photo-exposées. Pour les patients qui sexposent au soleil, des vêtements protecteurs, et lusage décran solaire (par exemple filtre minéral) peuvent être recommandés.
Les effets indésirables survenus plus fréquemment (≥ 3 %) dans le groupe erlotinib que dans le groupe placebo dans létude pivot BR.21 et chez au moins 10 % des patients du groupe erlotinib sont résumés par grade NCI-CTC (National Cancer Institute-Common Toxicity Criteria) dans le tableau 1.
La terminologie de la classification des effets indésirables en fonction de leur fréquence est la suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000) y compris les cas isolés.
Les effets indésirables sont présentés par ordre décroissant de gravité dans chaque catégorie de fréquence de survenue.
Tableau 1 : Effets indésirables (EI) très fréquents dans létude BR.21
|
|
Erlotinib N = 485 |
Placebo N = 242 |
||||
|
Grade NCI-CTC |
Tout grade |
3 |
4 |
Tout grade |
3 |
4 |
|
Terme préféré MedDRA |
% |
% |
% |
% |
% |
% |
|
Total des patients avec EI |
99 |
40 |
22 |
96 |
36 |
22 |
|
Infections et infestations Infection* |
24 |
4 |
0 |
15 |
2 |
0 |
|
Troubles du métabolisme et de la nutrition Anorexie |
52 |
8 |
1 |
38 |
5 |
< 1 |
|
Affections oculaires Kératoconjonctivite sèche Conjonctivite |
12 12 |
0 <1 |
0 0 |
3 2 |
0 <1 |
0 0 |
|
Affections respiratoires, thoraciques et médiastinales Dyspnée Toux |
41 33 |
17 4 |
11 0 |
35 29 |
15 2 |
11 0 |
|
Affections gastro-intestinales Diarrhée** Nausées Vomissements Stomatite Douleurs abdominales |
54 33 23 17 11 |
6 3 2 < 1 2 |
< 1 0 < 1 0 < 1 |
18 24 19 3 7 |
< 1 2 2 0 1 |
0 0 0 0 < 1 |
|
Affections de la peau et du tissu sous-cutané Eruption cutanée*** Prurit Sécheresse cutanée |
75 13 12 |
8 < 1 0 |
< 1 0 0 |
17 5 4 |
0 0 0 |
0 0 0 |
|
Troubles généraux et anomalies au site dadministration Asthénie |
52 |
14 |
4 |
45 |
16 |
4 |
*Les infections sévères, avec ou sans neutropénie ont inclus des cas de pneumopathie, de sepsis et de cellulite.
**Pouvant conduire à une déshydratation, une hypokaliémie et une insuffisance rénale.
***Eruption cutanée incluant la dermite acnéiforme.
Dans deux autres études de phase III randomisées en double aveugle contrôlées versus placebo B018192 (SATURN) et BO25460 (IUNO), lerlotinib était administré en maintenance après une première ligne de chimiothérapie. Ces études réalisées chez un total de 1 532 patients ayant un CBNPC avancé, récurrent ou métastatique après une première ligne de chimiothérapie standard à base de sels de platine, nont pas montré de nouveaux signaux de sécurité.
Les EI les plus fréquemment observés chez les patients traités par erlotinib dans les études BO18192 et BO25460 ont été des éruptions cutanées et des diarrhées (voir tableau 2). Aucune éruption cutanée ou diarrhée de grade 4 na été observée dans chacune de ces études. Dans létude BO18192, lerlotinib a été arrêté en raison déruptions cutanées et de diarrhées chez 1 % et < 1 % des patients respectivement alors que dans létude BO25460, aucune sortie détude en raison déruptions cutanées ou de diarrhées nest survenue. Une modification (arrêts ou réductions) de la posologie a été nécessaire en raison dune éruption cutanée ou dune diarrhée chez respectivement 8,3 % et 3 % des patients dans létude BO18192 et chez respectivement 5,6 % et 2,8 % des patients, dans létude BO25460.
Tableau 2 : Effets indésirables les plus fréquents dans les études BO18192 (SATURN) et BO25460 (IUNO)
|
|
BO18192 (SATURN)* |
BO25460 (IUNO)* |
||
|
|
Erlotinib n = 433 |
Placebo n = 445 |
Erlotinib n = 322 |
Placebo n = 319 |
|
|
% |
% |
% |
% |
|
Eruption cutanée, tous grades |
49,2 |
5,8 |
39,4 |
10,0 |
|
Grade 3 |
6,0 |
0 |
5,0 |
1,6 |
|
Diarrhée, tous grades |
20,3 |
4,5 |
24,2 |
4,4 |
|
Grade 3 |
1,8 |
0 |
2,5 |
0,3 |
*Analyse de la tolérance dans la population
Dans une étude de phase III, ML20650, randomisée en ouvert menée chez 154 patients traités par lerlotinib en première ligne de traitement chez des patients ayant un CBNPC et présentant des mutations activatrices de lEGFR, la tolérance a été évaluée chez 75 patients ; aucun nouveau signal de tolérance na été observé.
Les EI les plus fréquemment observés chez les patients traités par lerlotinib dans létude ML20650 ont été des éruptions cutanées et des diarrhées (tout grade, respectivement 80 % et 57 %), la plupart étaient de grade 1/2 et nont pas nécessité dintervention spécifique. Des éruptions cutanées et des diarrhées de grade 3 sont survenues chez respectivement 9 % et 4 % des patients. Aucune éruption cutanée ou diarrhée de grade 4 na été observée. Une sortie détude en raison déruptions cutanées ou de diarrhées est survenue chez 1 % des patients. Une modification (arrêt ou réduction) de la posologie en raison dune éruption cutanée ou dune diarrhée a été nécessaire chez respectivement 11 % et 7 % des patients.
Cancer du pancréas (erlotinib associé à la gemcitabine) :
Les effets indésirables les plus fréquents dans létude pivot PA.3, chez des patients atteints dun cancer du pancréas qui recevaient de lerlotinib 100 mg associé à la gemcitabine, ont été lasthénie, les éruptions cutanées et les diarrhées. Dans le groupe erlotinib plus gemcitabine, les éruptions cutanées et les diarrhées de grade 3/4 ont chacune été rapportées chez 5 % des patients. Les délais médians de survenue des éruptions cutanées et des diarrhées étaient respectivement de 10 et 15 jours. Les éruptions cutanées et les diarrhées ont chacune conduit à une réduction de la dose chez 2 % des patients et à une sortie détude allant jusquà 1 % des patients qui recevaient erlotinib plus gemcitabine.
Dans l'étude pivot PA.3, les effets indésirables survenus plus fréquemment (≥ 3 %) dans le groupe traité par erlotinib 100 mg plus gemcitabine que dans le groupe placebo plus gemcitabine et chez au moins 10 % des patients du groupe erlotinib 100 mg plus gemcitabine sont résumés par grades NCICTC (National Cancer Institute-Common Toxicity Criteria) dans le tableau 3.
La terminologie de la classification des effets indésirables en fonction de leur fréquence est la suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000) y compris les cas isolés.
Les effets indésirables sont présentés par ordre décroissant de gravité dans chaque catégorie de fréquence de survenue.
Tableau 3 : Effets indésirables (EI) très fréquents dans létude PA.3 (cohorte 100 mg)
|
|
Erlotinib N = 259 |
Placebo N = 256 |
||||
|
Grade NCI-CTC |
Tout Grade |
3 |
4 |
Tout Grade |
3 |
4 |
|
Terme préféré MedDRA |
% |
% |
% |
% |
% |
% |
|
Total des patients avec EI |
99 |
48 |
22 |
97 |
48 |
16 |
|
Infections et infestations Infection* |
31 |
3 |
< 1 |
24 |
6 |
< 1 |
|
Troubles du métabolisme et de la nutrition Perte de poids |
39 |
2 |
0 |
29 |
< 1 |
0 |
|
Affections psychiatriques Dépression |
19 |
2 |
0 |
14 |
< 1 |
0 |
|
Affections du système nerveux Neuropathie Céphalées |
13 15 |
1 < 1 |
< 1 0 |
10 10 |
< 1 0 |
0 0 |
|
Affections respiratoires, thoraciques et médiastinales Toux |
16 |
0 |
0 |
11 |
0 |
0 |
|
Affections gastro-intestinales Diarrhée** Stomatite Dyspepsie Flatulence |
48 22 17 13 |
5 < 1 < 1 0 |
< 1 0 0 0 |
36 12 13 9 |
2 0 < 1 < 1 |
0 0 0 0 |
|
Affections de la peau et du tissu sous-cutané Eruption cutanée*** Alopécie |
69 14 |
5 0 |
0 0 |
30 11 |
1 0 |
0 0 |
|
Troubles généraux et anomalies au site dadministration Asthénie Pyrexie Frissons |
73 36 12 |
14 3 0 |
2 0 0 |
70 30 9 |
13 4 0 |
2 0 0 |
*Les infections sévères, avec ou sans neutropénie, ont inclus des cas de pneumopathie, de sepsis et de cellulite.
**Pouvant conduire à une déshydratation, une hypokaliémie et une insuffisance rénale.
***Eruption cutanée incluant la dermite acnéiforme.
Autres observations :
Lévaluation de la tolérance de lerlotinib est basée sur les données obtenues chez plus de 1 500 patients ayant reçu au moins une dose de 150 mg derlotinib en monothérapie ainsi que chez plus de 300 patients ayant reçu de lerlotinib 100 ou 150 mg en association à la gemcitabine.
Les effets indésirables suivants ont été observés chez des patients ayant reçu de lerlotinib en monothérapie ou en association à une chimiothérapie.
Les effets indésirables très fréquents observés dans les études BR.21 et PA.3 sont présentés dans les tableaux 1 et 3 et les autres effets indésirables dont ceux observés dans les autres études sont résumés dans le tableau 4.
Les effets indésirables sont présentés par ordre décroissant de gravité dans chaque catégorie de fréquence de survenue.
Tableau 4 : Résumé des effets indésirables par catégorie de fréquence :
|
Classe de systèmes dorganes |
Très fréquent (≥ 1/10) |
Fréquent (≥ 1/100 à < 1/10) |
Peu fréquent (≥ 1/1 000 à < 1/100) |
Rare (≥ 1/10 000 à < 1/1 000) |
Très rare (< 1/10 000) |
|
Affections oculaires |
|
-Kératite -Conjonctivite1 |
-Modification des cils2 |
|
-Perforations de la cornée -Ulcérations de la cornée -Uvéite |
|
Affections respiratoires, thoraciques et médiastinales |
|
-Epistaxis -Affections Pulmonaires Interstitielles (API)3 graves |
|
|
|
|
Affections gastro-intestinales |
-Diarrhée7 |
-Hémorragies gastro-intestinales4, 7 |
-Perforations gastro-intestinales7 |
|
|
|
Affections hépatobiliaires |
-Anomalies des explorations fonctionnelles hépatiques5 |
|
|
-Insuffisance hépatique6 |
|
|
Affections de la peau et du tissu sous-cutané |
|
-Alopécie -Sécheresse cutanée1 -Paronychie -Folliculite -Acné/Dermatite acnéiforme -Fissures de la peau |
-Hirsutisme -Modification des sourcils -Ongles cassants et perte des ongles -Réactions cutanées légères telles que hyperpigmentation |
- Syndrome dérythrodysesthésie palmo-plantaire |
- Syndrome de Stevens-Johnson/syndrome de Lyell (nécrolyse épidermique toxique)7 |
|
Affections du rein et des voies urinaires |
|
-Insuffisance rénale1 |
-Néphrite1 -Protéinurie1 |
|
|
1 Dans létude PA.3.
2 Dont cils incarnés, pousse et épaississement excessif des cils.
3 Dont certaines fatales, chez des patients traités par erlotinib pour un CBNPC ou pour dautres tumeurs solides à un stade avancé (voir rubrique 4.4). Une incidence plus élevée a été observée chez les patients dorigine japonaise (voir rubrique 4.4).
4 Dans les études cliniques, certains cas ont été associés à l'administration conjointe de warfarine (voir rubrique 4.5) ou d'AINS.
5 Dont des augmentations de lalanine aminotransférase [ALAT], de laspartate aminotransférase [ASAT] et de la bilirubine. Ces anomalies ont été le plus souvent d'intensité légère ou modérée de survenue transitoire ou associées à des métastases hépatiques.
6 Certains cas ont été fatals. Des facteurs tels que des antécédents de troubles hépatiques ou des traitements hépatotoxiques concomitants ont été associés (voir rubrique 4.4).
7 Certains cas ont été fatals (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Symptômes
Des doses uniques derlotinib par voie orale allant jusquà 1000 mg derlotinib chez des volontaires sains et jusquà 1600 mg chez des patients atteints dun cancer ont été bien tolérées. Ladministration dune dose de 200 mg deux fois par jour a été mal tolérée par des volontaires sains au bout de seulement quelques jours de traitement. Les données issues de ces études indiquent que des effets indésirables sévères tels que diarrhées, éruptions cutanées et, possiblement augmentation de lactivité des aminotransférases hépatiques pourraient survenir au-delà de la dose recommandée.
Prise en charge
En cas de suspicion de surdosage, l'administration derlotinib doit être suspendue et un traitement symptomatique doit être instauré.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme daction
Lerlotinib est un inhibiteur de la tyrosine kinase du récepteur du facteur de croissance épidermique humain de type 1 (Epidermal Growth Factor Receptor (EGFR) également connu comme HER1). Lerlotinib est un puissant inhibiteur de la phosphorylation intracellulaire de lEGFR. LEGFR est exprimé à la surface de cellules normales et cancéreuses. Dans des modèles non cliniques, l'inhibition de la phosphotyrosine de lEGFR résulte en un arrêt de la prolifération et/ou à une mort cellulaire.
Des mutations de lEGFR peuvent conduire à une activation constitutive des voies de signalisation anti-apoptotique et de la prolifération. La puissante efficacité de l'erlotinib sur le blocage de la signalisation médiée par lEGFR dans ces tumeurs arborant des mutations positives de l'EGFR est attribuée à la liaison étroite de l'erlotinib au site de liaison de lATP dans le domaine de la kinase mutée de l'EGFR. En raison du blocage en aval de la signalisation, la prolifération des cellules est arrêtée, et la mort cellulaire est induite par la voie intrinsèque de l'apoptose. La régression de la tumeur est observée dans des modèles de souris où lexpression de ces mutations activatrices de lEGFR est renforcée.
Efficacité clinique
- Traitement en première ligne du cancer bronchique non à petites cellules (CBNPC) chez des patients présentant des mutations activatrices de lEGFR (erlotinib en monothérapie) :
Lefficacité derlotinib en première ligne de traitement des patients ayant un CBNPC présentant des mutations activatrices de lEGFR a été démontrée dans un essai de phase III, randomisé, en ouvert (ML20650, EURTAC). Cette étude a été conduite chez des patients caucasiens atteints dun CBNPC localement avancé ou métastatique (stades IIIB et IV) qui navaient reçu précédemment ni chimiothérapie ni traitement anticancéreux systémique pour leur maladie localement avancée et qui présentaient des mutations dans le domaine tyrosine kinase de lEGFR (délétion de lexon 19 ou mutation de lexon 21). Les patients ont été affectés par randomisation 1:1 à un traitement par erlotinib 150 mg ou 4 cycles de chimiothérapie à base de doublet de sel de platine.
Le critère principal était la survie sans progression (PFS) évaluée par linvestigateur. Les résultats d'efficacité sont résumés dans le tableau 5.
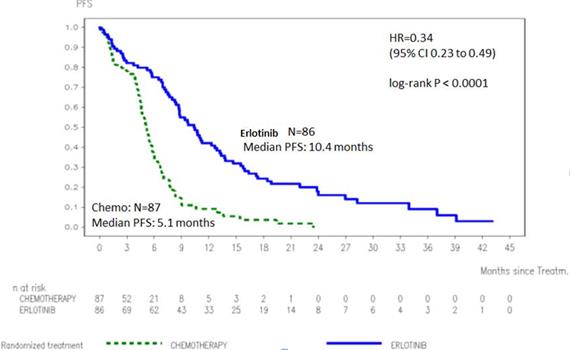
Figure 1 : Courbe de Kaplan-Meier de la PFS évaluée par linvestigateur dans lessai ML20650 (EURTAC) (cut-off davril 2012)
Tableau 5 : Résultats defficacité derlotinib versus chimiothérapie de lessai ML20650 (EURTAC)
|
|
|
Erlotinib |
Chimio-thérapie |
Hazard Ratio (IC à 95 %) |
Valeur de p |
|
Analyse intermédiaire planifiée (OS à 35 % de maturité) (n = 153) Date de Cut-off : Août 2010 |
|
n = 77 |
n = 76 |
|
|
|
Critère dévaluation primaire : Survie sans progression (PFS, médiane en mois)* Evaluée par linvestigateur ** Revue indépendante** |
9,4
10,4 |
5,2
5,4 |
0,42 [0,27-0,64] 0,47 [0,27-0,78] |
p < 0,0001
p = 0,003 |
|
|
Meilleur taux de réponse globale (RC/RP) |
54,5 % |
10,5 % |
|
p < 0,0001 |
|
|
Survie globale (OS) (mois) |
22,9 |
18,8 |
0,80 [0,47-1,37] |
p = 0,4170 |
|
|
Analyse exploratoire (OS à 40 % de maturité) (n = 173) Date de Cut-off : Janvier 2011 |
|
n = 86 |
n = 87 |
|
|
|
PFS (médiane en mois) Evaluée par linvestigateur |
9,7 |
5,2 |
0,37 [0,27-0,54] |
p < 0,0001 |
|
|
Meilleur taux de réponse globale (RC/RP) |
58,1 % |
14,9 % |
|
p < 0,0001 |
|
|
OS (mois) |
19,3 |
19,5 |
1,04 [0,65-1,68] |
p = 0,8702 |
|
|
Analyse actualisée (OS à 62 % de maturité) (n = 173) Date de Cut-off : Avril 2012 |
|
n = 86 |
n = 87 |
|
|
|
PFS (médiane en mois) |
10,4 |
5,1 |
0,34 [0,23-0,49] |
p < 0,0001 |
|
|
OS*** (mois) |
22,9 |
20,8 |
0,93 [0,64-1,36] |
p = 0,7149 |
RC = réponse complète ; RP = réponse partielle
* Une diminution de 58 % du risque de progression de la maladie ou de décès a été observée.
** Le taux de concordance entre lévaluation de linvestigateur et celle du comité de revue indépendant était de 70 %.
*** Un taux élevé de cross-over a été observé avec 82 % des patients du bras traité par chimiothérapie ayant reçu ultérieurement un inhibiteur de la tyrosine kinase de lEGFR, et tous ces patients excepté deux ayant reçu de lerlotinib.
- Traitement de maintenance du CBNPC après une première ligne de chimiothérapie (erlotinib en monothérapie) :
Lefficacité et la tolérance de lerlotinib dans le traitement de maintenance du CBNPC après une première ligne de chimiothérapie ont été étudiées dans un essai randomisé en double aveugle contrôlé versus placebo (B018192, SATURN). Cette étude a été conduite chez 889 patients atteints dun CBNPC localement avancé ou métastatique qui na pas progressé après 4 cycles de chimiothérapie à base de doublet de sel de platine. Les patients ont été affectés par randomisation 1:1 à un traitement par erlotinib 150 mg ou placebo, par voie orale, une fois par jour, jusquà progression de la maladie. Le critère principal de létude était la survie sans progression (Progression Free Survival : PFS) chez tous les patients. Les caractéristiques démographiques et pathologiques à linclusion des patients étaient bien équilibrées entre les deux bras de traitement. Les patients ayant un indice de performance ECOG PS > 1, des co-morbidités hépatiques ou rénales significatives, nétaient pas inclus dans létude.
Dans cette étude, lensemble de la population a montré un bénéfice pour le critère dévaluation principal qui était la PFS (risque relatif (Hazard Ratio : HR) = 0,71 p < 0,0001) et pour le critère dévaluation secondaire qui était la survie globale (overall survival : OS) (HR = 0,81 p = 0,0088). Cependant, le plus large bénéfice a été observé dans une analyse exploratoire prédéfinie chez des patients avec mutations activatrices de lEGFR (n = 49) en traduisant un bénéfice substantiel pour la PFS (HR = 0,10 ; IC à 95 % ; 0,04 à 0,25 ; p < 0,0001) et pour la survie globale avec un HR qui était de 0,83 (IC à 95 % ; 0,34 à 2,02). 67 % des patients du sous-groupe placebo avec la mutation EGFR positive ont reçu en seconde (ou plus tardive) ligne de traitement EGFR-TKIs.
Létude BO25460 (IUNO) a été menée chez 643 patients atteints dun CBNPC avancé sans mutation activatrice de lEGFR de la tumeur (délétion de lexon 19 ou mutation L858R de lexon 21) et qui na pas progressé après 4 cycles de chimiothérapie à base de platine.
Lobjectif de létude était de comparer la survie globale dune thérapie par erlotinib en première ligne de traitement de maintenance versus erlotinib administré au moment de la progression de la maladie. Létude na pas atteint son critère dévaluation principal. La survie globale de lerlotinib en première ligne de traitement de maintenance nétait pas supérieure au traitement erlotinib en seconde ligne de traitement chez les patients sans mutation activatrice de lEGFR de la tumeur (HR = 1,02 ; IC à 95 %; 0,85 à 1,22 ; p = 0,82). Le critère dévaluation secondaire PFS na pas montré de différence entre lerlotinib et le placebo en traitement de maintenance (HR = 0,94 ; IC à 95 %; 0,80 à 1,11 ; p = 0,48).
Sur la base des données de létude BO25460 (IUNO), lutilisation derlotinib nest pas recommandée en première ligne de traitement de maintenance chez les patients sans mutation activatrice de lEGFR.
- Traitement du CBNPC après échec dau moins un régime de chimiothérapie (erlotinib en monothérapie) :
Lefficacité et la tolérance de lerlotinib en traitement de deuxième/troisième ligne ont été démontrées dans un essai randomisé en double aveugle contrôlé versus placebo (BR.21) chez 731 patients atteints dun CBNPC localement avancé ou métastatique après échec d'au moins une ligne de chimiothérapie. Les patients ont été affectés par randomisation 2:1 à un traitement par erlotinib 150 mg ou placebo, par voie orale, une fois par jour. Les critères dévaluation de létude étaient notamment la survie globale, la survie sans progression (Progression Free Survival : PFS), le taux et la durée de réponse, le délai daggravation des symptômes liés au cancer du poumon (toux, dyspnée et douleurs), et la tolérance. Le critère principal de létude était la survie.
Les caractéristiques démographiques étaient bien équilibrées entre les deux groupes de traitement. Environ deux tiers des patients étaient de sexe masculin et l'indice de performance initial (Eastern Cooperative Oncology Group ECOG performance status (PS)) était de 2 chez environ un tiers des patients et de 3 chez 9 % des patients. Une chimiothérapie incluant un sel de platine avait été antérieurement administrée chez 93 % des patients du groupe erlotinib et chez 92 % des patients du groupe placebo, et respectivement 36 % et 37 % des patients avaient été traités par un taxane.
Le Risque Relatif (Hazard Ratio (HR)) ajusté de décès dans le groupe erlotinib par rapport au groupe placebo a été de 0,73 (IC à 95 % : 0,60 à 0,87) (p = 0,001). Le pourcentage de patients en vie à 12 mois a été de 31,2 % dans le groupe erlotinib et de 21,5 % dans le groupe placebo. La médiane de survie globale était de 6,7 mois dans le groupe erlotinib (IC à 95 % : 5,5 à 7,8 mois) comparée à 4,7 mois dans le groupe placebo (IC à 95 % : 4,1 à 6,3 mois).
Leffet sur la survie globale était exploré à travers différents sous-groupes de patients. Les effets derlotinib sur la survie globale étaient similaires chez les patients dont lECOG PS initial était de 2-3 (HR = 0,77 ; IC à 95 % : 0,6-1,0) ou de 0-1 (HR = 0,73 ; IC à 95 % : 0,6-0,9), les hommes (HR = 0,76 ; IC à 95 % : 0,6-0,9) ou les femmes (HR = 0,80 ; IC à 95 % : 0,6-1,1), les patients âgés de moins de 65 ans (HR = 0,75 ; IC à 95 % : 0,6-0,9) ou les patients plus âgés (HR = 0,79 ; IC à 95 % : 0,6-1,0), les patients ayant reçu auparavant un seul traitement de chimiothérapie (HR = 0,76, IC à 95 % : 0,6-1,0), ou plus de un traitement de chimiothérapie (HR = 0,75 ; IC à 95 % : 0,6-1,0), les patients Caucasiens (HR = 0,79 ; IC à 95 % : 0,6-1,0) ou Asiatiques (HR = 0,61 ; IC à 95 % : 0,4-1,0), les patients avec un adénocarcinome (HR = 0,71 ; IC à 95 % : 0,6-0,9) ou un carcinome épidermoïde (HR = 0,67 ; IC à 95 % : 0,5-0,9), mais pas chez les patients avec dautres types histologiques (HR 1,04 ; IC à 95 % : 0,7-1,5), les patients diagnostiqués au stade IV (HR = 0,92 ; IC à 95 % : 0,7-1,2) ou diagnostiqué à un stade < IV (HR = 0,65 ; IC à 95 % : 0,5-0,8). Le bénéfice derlotinib a été meilleur chez les patients nayant jamais fumé (HR survie : 0,42 ; IC à 95 % : 0,28-0,64) comparé aux fumeurs ou anciens fumeurs (HR = 0,87 ; IC à 95 % : 0,71-1,05).
Parmi les 45 % de patients dont le statut dexpression EGFR était connu, le Hazard Ratio était de 0,68 (IC à 95 % : 0,49-0,94) pour les patients avec des tumeurs EGFR-positif et de 0,93 (IC à 95 % : 0,63-1,36) pour les patients avec des tumeurs EGFR-négatif (déterminé par IHC en utilisant le kit EGFR pharmaDx et définissant le statut EGFR-négatif comme taux inférieur à 10 % des cellules tumorales colorées). Chez les 55 % de patients restants, dont le statut dexpression EGFR était inconnu, le HR était de 0,77 (IC à 95 % : 0,61- 0,98).
La survie médiane sans progression (PFS) était de 9,7 semaines dans le groupe erlotinib (IC à 95 % : 8,4 à 12,4 semaines) comparée à 8,0 semaines dans le groupe placebo (IC à 95 % : 7,9 à 8,1 semaines).
Le taux de réponse objective selon les critères RECIST (Response Evaluation Criteria in Solid Tumors) a été de 8,9 % (IC à 95 % : 6,4 à 12,0 %) dans le groupe erlotinib. Les 330 premiers patients ont été évalués de manière centralisée (taux de réponse : 6,2 %) ; 401 patients ont été évalués par les investigateurs (taux de réponse : 11,2 %).
La durée médiane de réponse a été de 34,3 semaines (allant de 9,7 à plus de 57,6 semaines). La proportion des patients ayant présenté une réponse complète ou partielle ou une stabilisation de la maladie a été de 44,0 % dans le groupe erlotinib et de 27,5 % dans le groupe placebo (p = 0,004).
Un bénéfice en survie a été également observé chez les patients traités par erlotinib n'ayant pas présenté une réponse tumorale objective (selon les critères RECIST). Cela a été montré avec un HR de décès de 0,82 (IC à 95 % : 0,68 à 0,99) chez les patients dont la meilleure réponse a été une stabilisation ou une progression de la maladie.
Lerlotinib a exercé un effet bénéfique en prolongeant significativement les délais daggravation de la toux, de la dyspnée et des douleurs comparativement au placebo.
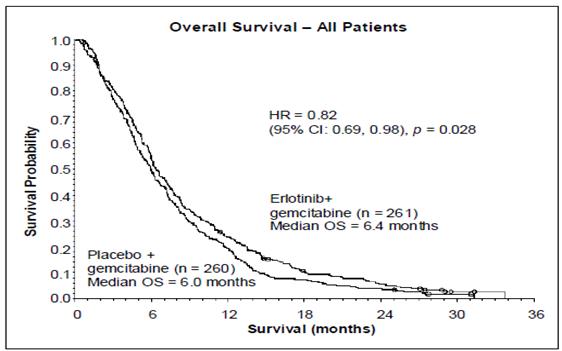
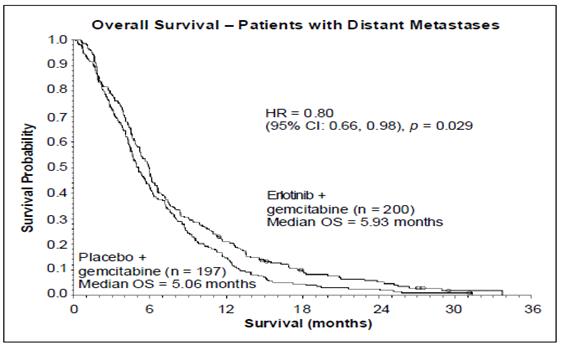
- Cancer du pancréas (erlotinib associé à la gemcitabine dans létude PA.3) :
Lefficacité et la tolérance de lerlotinib associé à la gemcitabine en traitement de première ligne ont été évaluées dans un essai randomisé, en double aveugle, contrôlé versus placebo chez des patients atteints d'un cancer du pancréas localement avancé, non résécable ou métastatique. Les patients ont été randomisés pour recevoir un traitement par erlotinib ou placebo une fois par jour en traitement continu plus gemcitabine IV (1000 mg/m², Cycle 1 - jours 1, 8, 15, 22, 29, 36 et 43 dun cycle de 8 semaines ; cycle 2 et cycles ultérieurs - jours 1, 8 et 15 dun cycle de 4 semaines [posologie et rythme dadministration de la gemcitabine approuvés dans le traitement du cancer du pancréas : voir le RCP de la gemcitabine]). Lerlotinib ou le placebo ont été pris par voie orale une fois par jour jusqu'à progression de la maladie ou apparition dune toxicité inacceptable. Le critère principal de létude était la survie globale.
Les caractéristiques démographiques et pathologiques à linclusion des patients étaient similaires entre les deux groupes de traitement, erlotinib 100 mg plus gemcitabine ou placebo plus gemcitabine, à lexception dune proportion légèrement plus élevée de femmes dans le groupe erlotinib/gemcitabine que dans le groupe placebo/gemcitabine :
|
A linclusion |
Erlotinib |
Placebo |
|
Femmes |
51 % |
44 % |
|
Indice de performance ECOG (PS) = 0 |
31 % |
32 % |
|
Indice de performance ECOG (PS) = 1 |
51 % |
51 % |
|
Indice de performance ECOG (PS) = 2 |
17 % |
17 % |
|
Maladie métastatique à linclusion |
77 % |
76 % |
La survie a été évaluée dans la population en intention de traiter sur la base des données obtenues lors du suivi de la survie. Les résultats sont présentés dans le tableau ci-dessous (les résultats du groupe de patients métastatique et localement avancé proviennent dune analyse exploratoire des sous-groupes).
|
Résultats |
Erlotinib (mois) |
Placebo (mois) |
Δ (mois) |
IC du Δ |
HR |
IC du HR |
Valeur de p |
|
Population globale |
|||||||
|
Médiane de survie globale |
6,4 |
6,0 |
0,41 |
-0,54-1,64 |
0,82 |
0,69-0,98 |
0,028 |
|
Moyenne de survie globale |
8,8 |
7,6 |
1,16 |
-0,05-2,34 |
|||
|
Population métastatique |
|||||||
|
Médiane de survie globale |
5,9 |
5,1 |
0,87 |
-0,26-1,56 |
0,80 |
0,66-0,98 |
0,029 |
|
Moyenne de survie globale |
8,1 |
6,7 |
1,43 |
0,17-2,66 |
|||
|
Population localement avancée |
|||||||
|
Médiane de survie globale |
8,5 |
8,2 |
0,36 |
-2,43-2,96 |
0,93 |
0,65-1,35 |
0,713 |
|
Moyenne de survie globale |
10,7 |
10,5 |
0,19 |
-2,43-2,69 |
|||


Dans une analyse post-hoc, les patients ayant un état clinique favorable à linclusion (faible intensité de douleur, bonne qualité de vie et bon indice de performance), peuvent tirer un meilleur bénéfice derlotinib. Le bénéfice est principalement lié à la présence de douleur de faible intensité.
Dans une analyse post-hoc, les patients sous erlotinib ayant développé une éruption cutanée avaient une survie globale plus longue que les patients nayant pas développé déruption cutanée (médiane de survie globale 7,2 mois contre 5 mois, risque relatif HR : 0,61). 90 % des patients sous erlotinib ont développé une éruption cutanée dans les 44 premiers jours. Le temps médian dapparition de léruption cutanée était de 10 jours.
Population pédiatrique
LAgence européenne des médicaments a accordé une dérogation à lobligation de soumettre les résultats détudes réalisées avec lerlotinib dans tous les sous-groupes de la population pédiatrique dans les indications du cancer bronchique non à petites cellules et du cancer du pancréas (voir rubrique 4.2 pour les informations concernant lusage pédiatrique).
5.2. Propriétés pharmacocinétiques
Absorption : Après administration orale, le pic de concentration plasmatique est obtenu après environ 4 heures. La biodisponibilité absolue a été estimée à 59 % dans une étude chez des volontaires sains. La prise daliments peut augmenter lexposition après une prise orale.
Distribution : La valeur moyenne du volume apparent de distribution de lerlotinib est de 232 litres. Lerlotinib diffuse dans les tissus tumoraux chez lhomme. Lors dune étude menée chez 4 patients, dont 3 atteints dun cancer bronchique non à petites cellules (CBNPC) et 1 d'un cancer du larynx, recevant une dose orale quotidienne de 150 mg derlotinib, des dosages effectués sur des prélèvements tumoraux obtenus par excision chirurgicale au 9ème jour de traitement ont indiqué des concentrations intra tumorales moyennes derlotinib de 1,185 ng/g de tissu, ce qui correspond en moyenne à 63 % (intervalle : 5 161 %) des concentrations plasmatiques maximales observées à létat déquilibre. Les principaux métabolites actifs étaient présents dans la tumeur à une concentration moyenne de 160 ng/g de tissu, soit globalement en moyenne 113 % (intervalle : 88 130 %) des concentrations plasmatiques maximales déterminées à létat déquilibre. La liaison aux protéines plasmatiques est denviron 95 %. Lerlotinib se lie à lalbumine sérique et à lalpha-1 glycoprotéine acide (α1GPA).
Biotransformation : Lerlotinib est métabolisé par les cytochromes hépatiques chez l'Homme, principalement par le CYP3A4 et, à un moindre degré, par le CYP1A2. Le métabolisme extra hépatique par le CYP3A4 intestinal, le CYP1A1 pulmonaire et le CYP1B1 du tissu tumoral contribuent potentiellement à la clairance métabolique de lerlotinib.
Trois voies métaboliques principales ont été identifiées : 1) O-déméthylation dune ou des deux chaînes latérales, suivie dune oxydation en acides carboxyliques ; 2) oxydation du groupement acétylène suivie dune hydrolyse en acide arylcarboxylique et 3) hydroxylation aromatique du groupement phénylacétylène. Des dosages in vitro et des études de modèles tumoraux in vivo ont montré que les principaux métabolites de lerlotinib, OSI-420 et OSI-413, produits par O-déméthylation de lune ou lautre des chaînes latérales exerçaient une activité similaire à celle de lerlotinib. Ils sont présents dans le plasma à des concentrations inférieures à 10 % de celles de lerlotinib et leurs paramètres pharmacocinétiques sont similaires à ce dernier.
Elimination : L'erlotinib est principalement excrété sous forme de métabolites dans les fèces (> 90 %), lélimination rénale ne représentant quune faible proportion (environ 9 %) dune dose administrée par voie orale. Moins de 2 % de la dose administrée oralement sont excrétés sous forme inchangée. Une analyse pharmacocinétique à l'échelon d'une population de 591 patients recevant lerlotinib en monothérapie a montré une clairance moyenne apparente de 4,47 l/h et une demi-vie médiane de 36,2 heures. De ce fait, le délai dobtention de létat déquilibre des concentrations plasmatiques devrait être voisin de 7-8 jours.
Pharmacocinétique dans des populations particulières :
En se basant sur les analyses de pharmacocinétique de population, aucune relation significative entre la clairance apparente prévue et lâge, le poids, le sexe et lorigine ethnique des patients n'a été observée. Les facteurs liés au patient et corrélés aux paramètres pharmacocinétiques de lerlotinib sont la bilirubinémie totale, la concentration en α-1GPA et être fumeur. Des valeurs augmentées des concentrations plasmatiques de la bilirubine totale et de la concentration en α-1GPA ont été associées à une diminution de la clairance de lerlotinib. La signification clinique de ces différences nest pas claire. Toutefois, la clairance de lerlotinib a été augmentée chez les fumeurs. Ceci a été confirmé par une étude pharmacocinétique chez des volontaires sains non-fumeurs ou fumeurs actifs traités par une dose orale unique de 150 mg derlotinib. La moyenne géométrique de la Cmax était de 1056 ng/mL chez les non-fumeurs et 689 ng/mL chez les fumeurs avec un rapport moyen de fumeurs à non-fumeurs de 65,2 % (IC à 95 % : 44,3 à 95,9, p = 0,031). La moyenne géométrique de lASC0-inf était de 18726 ngh/mL chez les non-fumeurs et 6718 ngh/mL chez les fumeurs avec un rapport moyen de 35,9 % (IC à 95 % : 23,7 à 54,3, p < 0,0001). La moyenne géométrique de la C24h était de 288 ng/mL chez les non-fumeurs et 34,8 ng/mL chez les fumeurs avec un rapport moyen de 12,1 % (IC à 95 % : 4,82 à 30,2, p = 0,0001).
Dans létude pivot de phase III dans le CBNPC, les fumeurs actifs ont atteints létat déquilibre de lerlotinib à une concentration plasmatique de 0,65 μg/mL (n = 16) ce qui correspond à une concentration environ 2 fois inférieure à celle danciens fumeurs ou ceux qui nont jamais fumés (1,28 μg/mL, n = 108). Cet effet était accompagné par une augmentation de 24 % de la clairance plasmatique apparente de lerlotinib. Dans une étude de phase I descalade de dose dans le CBNPC chez les patients fumeurs actifs, les analyses pharmacocinétiques à létat déquilibre ont montrés une augmentation dose dépendante de laire sous la courbe de lerlotinib lorsque la posologie derlotinib était augmentée de 150 mg à la dose maximale tolérée de 300 mg. Dans cette étude, létat déquilibre des concentrations plasmatiques à une posologie de 300 mg chez les fumeurs actifs était de 1,22 μg/mL (n = 17).
Du fait des résultats des études de pharmacocinétique lors du traitement par erlotinib, les patients fumeurs devront être encouragés à arrêter, compte tenu de la réduction possible des concentrations plasmatiques derlotinib.
Sur la base de létude de pharmacocinétique de population, il apparaît que la présence dun opioïde augmente lexposition denviron 11 %.
Une seconde analyse de pharmacocinétique de population a été menée et a intégré des données sur lerlotinib obtenues chez 204 patients atteints dun cancer du pancréas ayant reçu l'erlotinib en association à la gemcitabine. Cette analyse a démontré que les co-variables influençant la clairance de l'erlotinib chez les patients inclus dans létude menée dans le cancer du pancréas étaient similaires à celles observées lors de lanalyse pharmacocinétique précédente en monothérapie. Aucun nouvel effet de covariance n'a été identifié. L'administration conjointe de la gemcitabine ne modifie pas la clairance plasmatique de lerlotinib.
Population pédiatrique : Aucune étude na été spécifiquement menée en pédiatrie.
Population âgée : Aucune étude na été spécifiquement menée chez les personnes âgées.
Insuffisance hépatique : Lerlotinib est principalement éliminé par le foie. Chez les patients ayant des tumeurs solides et une insuffisance hépatique modérée (score de Child-Pugh 7-9), la moyenne géométrique de lASC0-t et la Cmax de lerlotinib étaient respectivement de 27000 ngh/mL et 805 ng/mL comparées à 29300 ngh/mL et 1090 ng/mL chez les patients ayant une fonction hépatique adéquate y compris ceux ayant un cancer primitif du foie ou des métastases hépatiques. Bien que la différence sur la Cmax soit statistiquement significative, cette différence nest pas considérée comme cliniquement pertinente. Aucune donnée nest disponible quant à linfluence de troubles fonctionnels hépatiques sévères sur les paramètres pharmacocinétiques de lerlotinib. Sur la base des analyses de pharmacocinétique de population, laugmentation des concentrations sériques en bilirubine totale était associée à une diminution de la clairance de lerlotinib.
Insuffisance rénale : Lerlotinib et ses métabolites ne sont pas excrétés de façon significative par voie rénale. Moins de 9 % dune dose unique sont éliminés dans les urines Sur la base des analyses de pharmacocinétique de population, aucune relation cliniquement significative na été observée entre la clairance de lerlotinib et la clairance de la créatinine. Mais, il ny a pas de données disponibles chez les patients ayant une clairance de la créatinine < 15 mL/min.
5.3. Données de sécurité préclinique
Du fait de son mode daction, l'erlotinib a un potentiel tératogène. Des données issues détudes de la toxicité sur la reproduction menées chez le rat et le lapin, à des doses voisines de la dose maximale tolérée et/ou toxiques pour les mères, ont reporté une toxicité de reproduction (embryotoxicité chez les rats, résorption embryonnaire et ftotoxicité chez les lapins) et une toxicité de développement (diminution de la croissance et de la survie chez les jeunes rats), mais nont révélé aucun signe de tératogénicité ou daltération de la fertilité. Ces résultats ont été observés à des expositions cliniquement significatives.
Les études de génotoxicité conventionnelles menées avec lerlotinib se sont révélées négatives. Des études de carcinogénicité de 2 ans réalisées chez des rats et des souris à des niveaux dexposition supérieurs au niveau dexposition thérapeutique humain (respectivement, jusquà 2 fois et 10 fois supérieurs en se basant sur la Cmax et/ou lASC) ont été négatives.
Une réaction cutanée phototoxique modérée a été observée chez les rats après irradiation par les UV.
Cellulose microcristalline, laurilsulfate de sodium, carboxyméthylamidon sodique, lactose monohydraté, silice colloïdale anhydre, stéarate de magnésium, huile végétale hydrogénée.
Pelliculage :
Lactose monohydraté, hypromellose, dioxyde de titane (E171), macrogol.
2 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Disponible en boîtes contenant 30 comprimés pelliculés sous plaquettes en PVC/Aclar/PVC/Aluminium ou 30 x 1 comprimé pelliculé sous plaquettes prédécoupées unitaires en PVC/Aclar/PVC//feuille daluminium.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Pas dexigences particulières pour lélimination.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
100-110 ESPLANADE DU GENERAL DE GAULLE
92931 PARIS LA DEFENSE CEDEX
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 300 831 1 6 : 30 comprimés pelliculés sous plaquettes (PVC/Aclar/PVC/Al).
· 34009 300 831 2 3 : (30 x 1) comprimés pelliculés sous plaquettes unitaires prédécoupées (PVC/Aclar/PVC/Al).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en oncologie ou en hématologie ou aux médecins compétents en cancérologie.