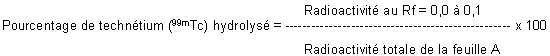
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 02/07/2008
OSTEOCIS. Trousse pour la préparation de la solution injectable d'oxidronate de technétium (99m Tc)
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Oxidronate de sodium (hydroxyméthylène disphosphonate de sodium, HMDP) ....................................... 3,0 mg
Pour un flacon.
Pour la liste complète des excipients, voir rubrique 6.1.
Le produit ne contient pas de conservateur antimicrobien.
Trousse pour préparation radiopharmaceutique.
Poudre pour solution injectable.
4.1. Indications thérapeutiques
Ce médicament est à usage diagnostique uniquement.
Après reconstitution avec une solution injectable de pertechnétate (99mTc) de sodium, la solution injectable d'oxidronate de technétium (99mTc) peut être utilisée comme produit de diagnostic pour la scintigraphie du squelette afin de détecter des zones dont l'ostéogenèse est anormale.
4.2. Posologie et mode d'administration
Chez l'adulte de masse corporelle de 50 à 70 kg, l'activité moyenne recommandée est de 500 MBq (300 à 700 MBq) administrée par voie intraveineuse stricte. Des activités différentes peuvent être justifiées.
Posologie pédiatrique
Chez l'enfant, l'activité à injecter est une fraction de celle recommandée chez l'adulte, fraction obtenue par l'application des coefficients multiplicateurs ci-dessous en fonction de la masse corporelle.
|
3 kg = 0,10 |
22 kg = 0,50 |
42 kg = 0,784 |
|
4 kg = 0,14 |
24 kg = 0,53 |
44 kg = 0, 80 |
|
6 kg = 0,19 |
26 kg = 0,56 |
46 kg = 0,82 |
|
8 kg = 0,23 |
28 kg = 0,58 |
48 kg = 0,85 |
|
10 kg = 0,27 |
30 kg = 0,62 |
50 kg = 0,88 |
|
12 kg = 0,32 |
32 kg = 0,65 |
52-54 kg = 0,90 |
|
14 kg = 0,36 |
34 kg = 0,68 |
56-58 kg = 0,92 |
|
16 kg = 0,40 |
36 kg = 0,71 |
60-62 kg = 0,96 |
|
18 kg = 0,44 |
38 kg = 0,73 |
64-66 kg = 0,98 |
|
20 kg = 0,46 |
40 kg = 0,76 |
68 kg = 0,99 |
Chez le très jeune enfant (jusqu'à 1an), une activité minimale de 40 MBq est nécessaire pour obtenir des images de qualité satisfaisante.
Les images acquises immédiatement après l'injection (par exemple dans le protocole dit en « trois phases »), ne reflètent qu'une partie de l'activité métabolique osseuse. La scintigraphie tardive est réalisée au plus tôt 2 heures après l'injection. Il serait souhaitable que le patient urine avant l'acquisition des images.
Hypersensibilité à la substance active ou à l'un des excipients.
4.4. Mises en garde spéciales et précautions d'emploi
Ce produit est un médicament radiopharmaceutique.
Les produits radiopharmaceutiques ne peuvent être réceptionnés, utilisés et administrés que par des personnes autorisées dans des services agréés. Les produits radiopharmaceutiques doivent être préparés de manière à satisfaire à la fois aux normes de radioprotection et de qualité pharmaceutique. Les précautions appropriées d'asepsie doivent être prises afin de satisfaire aux exigences des Bonnes Pratiques de Fabrication pharmaceutique.
Chez le jeune enfant la plaque de croissance épiphysaire fixe le produit et reçoit donc une irradiation supérieure à celle de l'os avoisinant.
Des précautions particulières doivent être prises pour éviter toute contamination par l'activité éliminée par les patients.
Pour diminuer la dose délivrée à la paroi de la vessie, le patient doit être convenablement hydraté et doit uriner fréquemment.
Afin de limiter la quantité de traceur présente dans la masse musculaire, tout exercice physique doit être évité entre l'injection du produit et la fin de l'examen.
Il faut prendre toute les précautions utiles lors de l'injection d'oxidronate de technétium (99mTc) afin d'éviter une administration sous-cutanée accidentelle qui pourrait provoquer une inflammation périvasculaire.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L'accumulation de l'oxidronate de technétium (99mTc) dans le squelette, et donc la qualité de l'examen scintigraphique, peut être limitée par des traitements comprenant des chélateurs, des tétracyclines ou d'autres médicaments contenant du fer.
L'administration régulière de médicaments contenant de l'aluminium, tels que antiacides, peut provoquer une accumulation particulièrement élevée de technétium (99mTc) dans le foie, probablement due à la formation de colloïdes marqués.
Lorsqu'il est nécessaire d'administrer des produits radiopharmaceutiques à la femme en âge de procréer, toute éventualité de grossesse doit être écartée. Toute femme n'ayant pas eu ses règles doit être considérée comme enceinte jusqu'à preuve du contraire. Dans le doute, il est important que l'exposition aux radiations soit réduite au minimum pour obtenir les informations cliniques souhaitées. D'autres techniques n'impliquant pas l'emploi de radiations ionisantes doivent être envisagées.
Les examens utilisant des radionucléides chez la femme enceinte entraînent également l'irradiation du ftus. Il ne faut réaliser au cours de la grossesse que les seules investigations absolument nécessaires lorsque le bénéfice probable dépasse les risques encourus par la mère et le ftus.
L'administration de 700 MBq d'oxidronate de technétium (99mTc) à une patiente ayant une fixation osseuse normale, entraîne une dose absorbée par l'utérus de 4,27 mGy. Cette dose décroît à 2,03 mGy chez les patientes à forte fixation osseuse et/ou à fonction rénale réduite.
En cas d'allaitement, si l'administration de ce produit est indispensable, le lait doit être tiré avant l'injection et conservé pour être utilisé ultérieurement. L'allaitement doit être suspendu pendant au moins 4heures après l'injection et le lait produit pendant cette période doit être éliminé.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés.
Pour tous les patients, l'exposition aux radiations ionisantes doit être justifiée par le bénéfice diagnostique attendu. L'activité administrée doit correspondre à la plus faible dose de radiation possible compatible avec l'obtention de l'information diagnostique escomptée.
L'exposition aux radiations ionisantes peut potentiellement induire des cancers ou développer des déficiences héréditaires. L'expérience montre que pour ce qui est des examens diagnostiques en médecine nucléaire, la fréquence de ces effets indésirables est très faible en raison des faibles activités utilisées.
Pour la plupart des examens de médecine nucléaire à des fins de diagnostic, la dose de radiations délivrée (E: dose efficace) est inférieure à 20 mSv.
Après l'administration de l'oxidronate de technétium (99mTc) les effets indésirables sont extrêmement rares (inférieurs à 1 sur 200 000 administrations): réactions anaphylactiques, rougeur, nausées, hypotension et parfois arthralgies. Ces symptômes peuvent apparaître 4 à 24 heures après l'administration.
En cas d'administration d'une activité excessive d'oxidronate de technétium (99mTc), la dose délivrée au patient doit être réduite en augmentant autant que possible l'élimination du radionucléide par une diurèse forcée avec mictions fréquentes.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique:produit radiopharmaceutique à usage diagnostique,
Code ATC:V09BA01.
L'oxidronate de technétium (99mTc), aux concentrations et aux activités utilisées pour les examens diagnostiques, ne semble pas exercer d'effet pharmacodynamique.
5.2. Propriétés pharmacocinétiques
Après l'administration intraveineuse, l'oxidronate de technétium (99mTc) est rapidement distribué dans l'espace extra-cellulaire. La fixation par l'os est presque immédiate et progresse rapidement. 30 minutes après l'injection, 10 % de l'activité injectée est encore présente dans la circulation sanguine. Cette valeur décroît à 5 %, 3 %, 1, 5%, et 1 % respectivement 1, 2, 3 et 4 heures après l'injection. L'élimination se fait par voie urinaire.
Environ 30 % de l'activité administrée sont éliminées pendant la première heure, 48 % dans les 2 heures et 60 % dans les 6 heures.
5.3. Données de sécurité préclinique
Ce produit n'est pas destiné à être administré de façon régulière ou continue. Des études sur la reproduction, sur la mutagenèse ou la carcinogenèse à long terme n'ont pas été entreprises.
Des anomalies hépatiques minimes ont été observées chez le rat après administration de 30 mg/kg.
Lors d'études de toxicité subaiguë, des doses de 10 mg/kg/jour pendant 14 jours n'ont pas donné de réaction chez le rat, par contre des changements histologiques au niveau du foie (microgranulomes) ont été observés chez le chien après l'administration de 3 et 10 mg/kg/jour pendant 14 jours. En outre, chez les chiens traités durant 14 jours consécutifs, on observe des indurations persistantes au point d'injection.
Chlorure stanneux dihydraté, acide ascorbique, chlorure de sodium, sous atmosphère d'azote.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
1 an.
8 heures après marquage pour le produit marqué.
6.4. Précautions particulières de conservation
Conserver la trousse et le produit marqué au réfrigérateur (entre 2°C et 8°C).
Le stockage des médicaments radiopharmaceutiques doit être effectué conformément aux réglementations nationales relatives aux produits radioactifs.
6.5. Nature et contenu de l'emballage extérieur
Poudre en flacon de 15 mL (verre étiré incolore de type I), muni d'un bouchon (caoutchouc) scellé par une capsule (aluminium).
Boîte de 5 flacons multidoses.
6.6. Précautions particulières délimination et de manipulation
L'administration de produits radiopharmaceutiques présente des risques pour l'entourage du patient en raison de l'irradiation externe ou de la contamination par les urines, les vomissements, les expectorations. Par conséquent, il faut prendre des mesures de protection contre les radiations conformément aux réglementations nationales.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
CIS BIO INTERNATIONAL
RN 306
BP 32
91192 Gif-sur-Yvette Cedex
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 554 212-5: 14,2 mg de lyophilisat en flacon (verre), boîte de 5 flacons.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter par le titulaire]
Le technétium (99mTc) décroît en émettant principalement un rayonnement gamma d'une énergie de 140 keV, avec une période de 6 heures, en donnant du technétium-99 pouvant être considéré comme un isotope stable.
Selon les publications 53, 60 et 80 de la CIPR (Commission Internationale pour la Protection Radiologique), les doses de radiation absorbées par les patients sont les suivantes:
Selon CIPR 80
Fixation osseuse normale
|
Organe |
Dose absorbée par unité d'activité administrée (en mGy/MBq) |
||||
|
|
Adulte |
15 ans |
10 ans |
5 ans |
1 an |
|
Surfaces osseuses |
0,063 |
0,082 |
0,13 |
0,22 |
0,53 |
|
Paroi vésicale |
0,048 |
0,060 |
0,088 |
0,073 |
0,13 |
|
Moelle osseuse |
0,0092 |
0,010 |
0,017 |
0,033 |
0,067 |
|
Reins |
0,0073 |
0,0088 |
0,012 |
0,018 |
0,032 |
|
Utérus |
0,0063 |
0,0076 |
0,012 |
0,011 |
0,018 |
|
Paroi côlon descendant |
0,0038 |
0,0047 |
0,0072 |
0,0075 |
0,013 |
|
Ovaires |
0,0036 |
0,0046 |
0,0066 |
0,0070 |
0,012 |
|
Colon |
0,0027 |
0,0034 |
0,0053 |
0,0061 |
0,011 |
|
Testicules |
0,0024 |
0,0033 |
0,0055 |
0,0058 |
0,011 |
|
Intestin grêle |
0,0023 |
0,0029 |
0,0044 |
0,0053 |
0,0095 |
|
Surrénales |
0,0021 |
0,0027 |
0,0039 |
0,0058 |
0,011 |
|
Paroi côlon ascendant |
0,0019 |
0,0024 |
0,0039 |
0,0051 |
0,0089 |
|
Autres tissus |
0,0019 |
0,0023 |
0,0034 |
0,0045 |
0,0079 |
|
Muscles |
0,0019 |
0,0023 |
0,0034 |
0,0044 |
0,0079 |
|
Cerveau |
0,0017 |
0,0021 |
0,0028 |
0,0043 |
0,0061 |
|
Pancréas |
0,0016 |
0,0020 |
0,0031 |
0,0045 |
0,0082 |
|
Vésicule biliaire |
0,0014 |
0,0019 |
0,0035 |
0,0042 |
0,0067 |
|
Rate |
0,0014 |
0,0018 |
0,0028 |
0,0045 |
0,0079 |
|
Poumons |
0,0013 |
0,0016 |
0,0024 |
0,0036 |
0,0068 |
|
Thyroïde |
0,0013 |
0,0016 |
0,0023 |
0,0035 |
0,0056 |
|
Foie |
0,0012 |
0,0016 |
0,0025 |
0,0036 |
0,0066 |
|
Cur |
0,0012 |
0,0016 |
0,0023 |
0,0034 |
0,0060 |
|
Paroi de l'estomac |
0,0012 |
0,0015 |
0,0025 |
0,0035 |
0,0066 |
|
Peau |
0,0010 |
0,0013 |
0,0020 |
0,0029 |
0,0055 |
|
sophage |
0,0010 |
0,0013 |
0,0019 |
0,0030 |
0,0053 |
|
Thymus |
0,0010 |
0,0013 |
0,0019 |
0,0030 |
0,0053 |
|
Seins |
0,00071 |
0,00089 |
0,0014 |
0,0022 |
0,0042 |
|
Dose efficace (mSv/MBq) |
0,0057 |
0,0070 |
0,011 |
0,014 |
0,027 |
Pour ce produit, la dose efficace (E) pour un individu de 70 kg résultant de l'administration d'une activité de 700 MBq est d'environ 4,0 mSv.
Après l'administration d'une activité de 700 MBq, la dose absorbée par les organes cibles (os) est de 44,1 mGy, et de 33,6 mGy pour l'organe critique (paroi de la vessie).
Selon CIPR 53 et 60
Fixation osseuse élevée et/ou insuffisance rénale sévère
|
Organe |
Dose absorbée par unité d'activité administrée (en mGy/MBq) |
||||
|
|
Adulte |
15 ans |
10 ans |
5 ans |
1 an |
|
Surfaces osseuses |
0,12 |
0,16 |
0,26 |
0,43 |
1,0 |
|
Moelle osseuse |
0,018 |
0,023 |
0,037 |
0,072 |
0,14 |
|
Surrénales |
0,0035 |
0,0050 |
0,0072 |
0,011 |
0,021 |
|
Paroi côlon descendant |
0,0034 |
0,0042 |
0,0065 |
0,0096 |
0,018 |
|
Pancréas |
0,0032 |
0,0040 |
0,0059 |
0,0089 |
0,016 |
|
Intestin grêle |
0,0031 |
0,0038 |
0,0057 |
0,0085 |
0,016 |
|
Reins |
0,0030 |
0,0037 |
0,0056 |
0,0087 |
0,016 |
|
Poumons |
0,0030 |
0,0037 |
0,0053 |
0,0081 |
0,015 |
|
Autres tissus |
0,0030 |
0,0036 |
0,0053 |
0,0081 |
0,015 |
|
Paroi côlon ascendant |
0,0029 |
0,0036 |
0,0053 |
0,0086 |
0,015 |
|
Ovaires |
0,0029 |
0,0041 |
0,0059 |
0,0089 |
0,016 |
|
Utérus |
0,0029 |
0,0037 |
0,0054 |
0,0082 |
0,015 |
|
Foie |
0,0027 |
0,0033 |
0,0049 |
0,0075 |
0,014 |
|
Paroi de l'estomac |
0,0026 |
0,0032 |
0,0051 |
0,0073 |
0,014 |
|
Rate |
0,0026 |
0,0034 |
0,0051 |
0,0078 |
0,015 |
|
Paroi vésicale |
0,0025 |
0,0035 |
0,0054 |
0,0074 |
0,015 |
|
Thyroïde |
0,0024 |
0,0037 |
0,0054 |
0,0083 |
0,014 |
|
Testicules |
0,0023 |
0,0027 |
0,0039 |
0,0060 |
0,011 |
|
Seins |
0,0021 |
0,0021 |
0,0032 |
0,0051 |
0,0096 |
|
Dose efficace (mSv/MBq) |
0,0053 |
0,0065 |
0,011 |
0,020 |
0,041 |
En cas de fixation osseuse élevée et d'insuffisance rénale sévère, la dose efficace résultant de l'administration d'une activité de 700 MBq d'oxidronate de technétium (99mTc) est de 3,7 mSv. La dose absorbée par les organes cibles (os) est de 84 mGy, et de 12,6 mGy pour l'organe critique (moelle érythropoïétique).
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Le produit ne contient pas de conservateur antimicrobien.
Les précautions appropriées d'asepsie et de radioprotection doivent être respectées.
Méthode de préparation
Placer un flacon de la trousse dans une protection de plomb appropriée.
Le flacon ne doit jamais être ouvert. Après désinfection du bouchon, à l'aide d'une seringue hypodermique, introduire à travers le bouchon 2 à 10 ml de solution injectable stérile et apyrogène de pertéchnétate (99mTc) de sodium. L'activité varie en fonction du volume de 0,75 à 11 GBq.
La solution de pertechnétate (99mTc) de sodium doit être conforme aux spécifications de la Pharmacopée Européenne.
Ne pas utiliser d'aiguille de mise à l'air car le contenu du flacon est sous azote: après introduction du volume requis de pertechnétate (99mTc) de sodium, prélever, sans retirer l'aiguille du bouchon, un volume équivalent d'azote afin d'éviter toute surpression dans le flacon.
Agiter régulièrement pendant 2 minutes et laisser reposer 15 minutes.
Le flacon doit être conservé à l'intérieur de sa protection de plomb.
Après désinfection du bouchon, la solution doit être prélevée aseptiquement à travers le bouchon à l'aide d'une seringue stérile, protégée par un étui de plomb, et d'une aiguille stérile.
Contrôle de qualité.
La solution obtenue doit être incolore, limpide et de pH compris entre 5,0 et 7,0.
La limpidité de la solution après préparation, le pH et l'activité doivent être vérifiés avant utilisation.
Pureté radiochimique
La qualité du marquage peut être vérifiée selon la procédure suivante:
Méthode
Chromatographie sur papier.
Matériel et réactifs
· Feuilles à chromatographie: 2 feuilles de papier à chromatographie Whatman 17 Chr (A et B). Tracer à 2,5 cm de l'une des extrémités de chaque feuille une fine ligne dite "ligne de dépôt".
· Phases mobiles
o Solvant A: Solution de chlorure de sodium à 0,9 %.
o Solvant B: Mélange méthanol / eau (85/15 V/V)
· Cuves à chromatographie
o 2 cuves à chromatographie de verre (A et B), dont les dimensions sont en rapport avec celles des feuilles à utiliser, munies d'un couvercle assurant une fermeture étanche. Maintenir les récipients fermés.
· Divers
o Pinces, ciseaux, seringues, aiguilles, unité de comptage appropriée.
Procédure
Ne pas laisser l'air pénétrer dans le flacon à tester et maintenir tous les flacons contenant une solution radioactive dans une protection de plomb.
· Introduire respectivement dans les cuves à chromatographie A et B un volume approprié de phases mobiles A et B tel que l'épaisseur de la couche obtenue soit de 2 cm.
· A l'aide d'une seringue munie d'une aiguille, déposer sur la «ligne de dépôt» de chacune des feuilles (A et B) une goutte de la préparation à examiner.
· A l'aide des pinces, introduire verticalement une feuille dans chacune des cuves à chromatographie, « lignes de dépôt » vers le bas. Fermer les cuves.
· Quand le solvant a atteint l'extrémité, retirer chaque feuille à l'aide de pinces et laisser sécher à l'air.
· Identifier les deux feuilles et les couper aux Rf = 0,15 et Rf = 0,4.
· Compter séparément chaque portion et noter les valeurs obtenues. Le temps de comptage doit être constant et le bruit de fond doit être soustrait.
· Calculs
Calculer le pourcentage de technétium (99mTc) hydrolysé:
Calculer le pourcentage de technétium (99mTc) libre:

Calculer le pourcentage de technétium (99mTc) lié:
Pourcentage de technétium (99mTc) lié = 100 % - (% technétium (99mTc) hydrolysé + % technétium (99mTc) libre).
Spécifications
Le pourcentage de technétium (99mTc) lié doit être supérieur à 95 % et le pourcentage de la somme: technétium (99mTc) hydrolysé + technétium (99mTc) libre doit être inférieur à 5 %.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament réservé à l'usage hospitalier.
Les produits radiopharmaceutiques ne doivent être utilisés que par des personnes qualifiées. Ils ne peuvent être délivrés qu'à des praticiens ayant obtenu l'autorisation spéciale prévue à l'article R 1333-24 du code de la Santé Publique.