RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 16/07/2015
SAYANAPRESS 104 mg,suspension injectable en injecteur prérempli
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Acétate de médroxyprogestérone 104 mg
pour 0,65 ml de suspension injectable
Chaque injecteur unidose prérempli contient 104 mg dacétate de médroxyprogestérone (AMP).
Excipients à effet notoire :
Parahydroxybenzoate de méthyle (E218) 1,04 mg pour 0,65 ml
Parahydroxybenzoate de propyle (E216) 0,0975 mg pour 0,65 ml
Sodium 2,47 mg pour 0,65 ml
Pour la liste complète des excipients, voir rubrique 6.1.
Suspension injectable en injecteur prérempli.
Suspension homogène de couleur blanche à blanc cassé.
4.1. Indications thérapeutiques
En raison de la possibilité dune perte de densité minérale osseuse (DMO) chez les femmes de tous âges qui prennent SAYANAPRESS à long terme (voir rubrique 4.4), une évaluation du rapport bénéfices/risques, tenant compte également de la diminution de la DMO qui survient au cours de la grossesse et/ou lallaitement, doit être envisagée avant ladministration de SAYANAPRESS.
Il est aussi important que la patiente soit informée de la nature prolongée des effets de ce produit, notamment du retour retardé de la fertilité (voir rubrique 4.4).
Utilisation chez les adolescentes (12-18 ans)
Chez les adolescentes, lutilisation de SAYANAPRESS nest indiquée que lorsque les autres méthodes de contraception sont jugées inadaptées ou inacceptables, en raison des effets inconnus à long terme de la perte osseuse associée à SAYANAPRESS au cours de la période critique de croissance osseuse (voir rubrique 4.4).
SAYANAPRESS na pas été étudié chez les femmes de moins de 18 ans mais des données, pour des injections de lAMP par voie intramusculaire, sont disponibles dans cette population.
4.2. Posologie et mode d'administration
Le récipient unidose de SAYANAPRESS doit être agité vigoureusement avant utilisation pour sassurer que la dose à administrer se présente sous forme dune suspension homogène. Le contenu est hermétiquement scellé dans le réservoir de linjecteur. Linjecteur doit être activé avant lutilisation. La procédure dactivation consiste à percer le joint interne de façon à ce que le médicament puisse passer dans laiguille lorsque le réservoir est sous pression. Le liquide ne remplit pas complètement le réservoir. Une petite bulle dair se trouve au-dessus du liquide. Lors de ladministration, linjecteur doit être utilisé avec laiguille orientée vers le bas. Ceci garantit que la dose complète de liquide est délivrée à travers laiguille. Le médicament doit être injecté lentement pendant 5 à 7 secondes environ. Le traitement doit être instauré par un médecin ou tout autre professionnel de santé habilité à prescrire SAYANAPRESS puis administré par injection sous-cutanée (SC) dans la partie antérieure de la cuisse ou dans labdomen.
Pour des instructions détaillées concernant la préparation et linjection de SAYANAPRESS, voir la rubrique 6.6 « Précautions particulières délimination et de manipulation » et la rubrique « Informations réservées aux professionnels de santé » de la notice.
Posologie
Adultes
Première injection : Pour obtenir une couverture contraceptive pendant le premier cycle dutilisation, une injection SC de 104 mg doit être administrée pendant les cinq premiers jours dun cycle menstruel normal. Si linjection est pratiquée selon ces instructions, aucune autre mesure contraceptive nest requise.
Doses suivantes : A partir de la deuxième injection, le traitement doit être administré à intervalles de 13 semaines ; tant que linjection est pratiquée dans un délai nexcédant pas sept jours après cette période, aucune autre mesure contraceptive (par exemple, méthode barrière) nest nécessaire. Si le délai écoulé depuis linjection précédente est supérieur à 14 semaines (13 semaines plus 7 jours), quelle quen soit la raison, léventualité dune grossesse doit être écartée avant ladministration de linjection suivante. Lefficacité de SAYANAPRESS repose sur le respect du schéma thérapeutique recommandé.
Post-partum : Si la patiente nallaite pas, linjection doit être administrée dans les 5 jours suivant laccouchement (pour sassurer autant que possible que la patiente ne soit pas enceinte). Si linjection est administrée à un autre moment, léventualité dune grossesse doit être écartée.
Si la patiente allaite, linjection ne doit pas être administrée avant 6 semaines après laccouchement, lorsque le système enzymatique du nourrisson est plus développé (voir rubrique 4.6).
Des données indiquent que les femmes ayant pris SAYANAPRESS immédiatement après laccouchement peuvent présenter des saignements prolongés et abondants. Cest pourquoi, le médicament doit être utilisé avec précaution pendant la période puerpérale. Les femmes qui envisagent dutiliser le produit immédiatement après un accouchement ou une interruption de grossesse doivent être informées du risque accru de saignements abondants ou prolongés. Il est rappelé aux médecins que lovulation peut se produire dès la quatrième semaine chez la patiente en post-partum qui nallaite pas.
Changement de méthodes de contraception : Lors du changement de méthode de contraception pour SAYANAPRESS, ce médicament doit être administré de façon à offrir une couverture contraceptive continue en fonction du mécanisme daction des deux méthodes (par exemple, les patientes qui passent dun contraceptif oral à SAYANAPRESS doivent recevoir leur première injection dans les 7 jours suivant la prise du dernier comprimé actif du contraceptif oral).
Populations particulières
Patientes souffrant dinsuffisance hépatique : Leffet dune maladie hépatique sur la pharmacocinétique de SAYANAPRESS nest pas connu. SAYANAPRESS étant largement éliminé par voie hépatique, il est possible quil soit faiblement métabolisé chez les patientes souffrant dinsuffisance hépatique sévère (voir rubrique 4.3).
Patientes souffrant dinsuffisance rénale : Leffet dune maladie rénale sur la pharmacocinétique de SAYANAPRESS nest pas connu. Aucun ajustement posologique ne devrait être nécessaire chez les femmes en insuffisance rénale, puisque SAYANAPRESS est presque exclusivement éliminé par métabolisme hépatique.
Population pédiatrique
SAYANAPRESS nest pas indiqué avant lapparition des premières règles (voir rubrique 4.1). Des données chez les adolescentes (12-18 ans) sont disponibles en ce qui concerne ladministration IM dAMP (voir rubriques 4.4 et 5.1). Hormis les préoccupations relatives à la perte de DMO, la sécurité et lefficacité de SAYANAPRESS devraient être similaires chez les adolescentes réglées et chez les femmes adultes.
Lacétate de médroxyprogestérone est contre-indiqué :
· chez les patientes qui ont une hypersensibilité connue à lAMP ou à lun des excipients mentionnés à la rubrique 6.1
· en cas de grossesse confirmée ou suspectée
· chez les femmes qui présentent une tumeur maligne confirmée ou suspectée du sein ou des organes génitaux
· chez les patientes qui présentent des saignements vaginaux non diagnostiqués
· chez les patientes qui souffrent dinsuffisance hépatique sévère
· chez les patientes qui souffrent dune maladie osseuse métabolique
· chez les patientes qui souffrent dune maladie thromboembolique et les patientes qui présentent ou qui ont des antécédents de maladie cérébrovasculaire.
4.4. Mises en garde spéciales et précautions d'emploi
Mises en garde spéciales
Perte de Densité Minérale Osseuse :
Cette perte de DMO est particulièrement préoccupante pendant ladolescence et au début de lâge adulte, une période critique de croissance osseuse. On ignore si lutilisation dAMPR en injection sous-cutanée chez des femmes plus jeunes diminuera le pic de masse osseuse et augmentera le risque de fracture par la suite.
Une étude visant à évaluer les effets sur la DMO de lacétate de médroxyprogestérone par voie IM (DEPO-PROVERA, AMPR) chez des adolescentes a montré que son utilisation était associée à une diminution significative de la DMO par rapport aux valeurs initiales. Chez un petit nombre de femmes qui étaient suivies, la DMO moyenne se rapprochait à peu près des valeurs initiales dans les 1 à 3 ans suivant larrêt du traitement. Chez les adolescentes, SAYANAPRESS peut être utilisé mais uniquement après discussion avec la patiente des autres méthodes de contraception et que celles-ci aient été jugées inadaptées ou inacceptables.
Chez les femmes de tous âges, une réévaluation minutieuse des risques et des bénéfices du traitement doit être effectuée chez celles qui souhaitent poursuivre le traitement au-delà de 2 ans. En particulier, chez les femmes qui présentent des facteurs de risque significatifs dostéoporose de par leur mode de vie ou leur état de santé, dautres méthodes de contraception doivent être envisagées avant dutiliser SAYANAPRESS.
Les facteurs de risque significatifs dostéoporose comprennent :
· Labus dalcool et/ou le tabagisme
· Lutilisation chronique de médicaments pouvant réduire la masse osseuse, comme les anticonvulsivants ou les corticoïdes
· Un indice de masse corporel bas ou des troubles alimentaires, comme lanorexie mentale ou la boulimie
· Des antécédents de fracture secondaire à un traumatisme mineur
· Des antécédents familiaux dostéoporose
Une étude de cohorte rétrospective utilisant les données de la General Practice Research Database (GPRD) a rapporté que les femmes qui reçoivent des injections dAMP (AMPR) présentent un risque plus élevé de fractures, comparativement aux utilisatrices de contraceptifs qui nont pas pris dAMPR (ratio du taux dincidence : 1,41 ; IC à 95 % : 1,35-1,47 pour la période de suivi de cinq ans) ; on ignore si ce phénomène est dû à lutilisation dAMPR ou à dautres facteurs en lien avec le mode de vie qui influent sur le taux de fractures. En revanche, chez les femmes qui reçoivent lAMPR, le risque de fractures avant et après le début du traitement par AMPR nest pas majoré (risque relatif : 1,08 ; IC à 95 % : 0,92-1,26). Il est important de noter que cette étude na pas pu déterminer si lutilisation dAMPR avait un effet sur le taux de fractures plus tard au cours de la vie.
Pour plus dinformations sur les fluctuations de la DMO chez les femmes adultes et adolescentes, comme cela a été rapporté dans des études cliniques récentes, se référer à la rubrique 5.1 (Propriétés pharmacodynamiques). Il est important que les femmes de tous âges prennent des quantités adéquates de calcium et de vitamine D pour préserver leur santé osseuse, que ce soit par le biais de lalimentation ou dune supplémentation,.
Menstruations irrégulières :
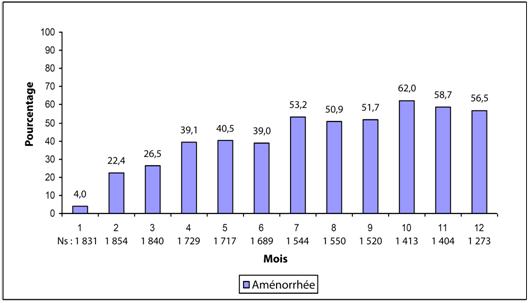
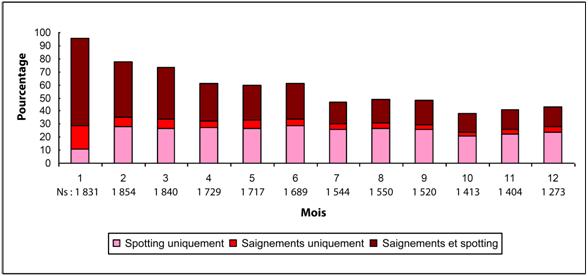
La plupart des femmes ayant utilisé lAMPR en injection sous-cutanée ont présenté une modification de leur cycle menstruel. Les patientes doivent être correctement informées sur la possibilité de troubles menstruels et dun éventuel retard du retour de l'ovulation. Avec la poursuite des injections sous-cutanées dAMPR, moins de femmes ont présenté des saignements irréguliers et plus de femmes ont présenté une aménorrhée. Après avoir reçu leur quatrième dose, 39 % des femmes ont développé une aménorrhée au cours du sixième mois. Au douzième mois, 56,5 % des femmes ont développé une aménorrhée. Les modifications du cycle menstruel observées dans les trois études évaluant leffet contraceptif sont présentées dans les figures 1 et 2. La figure 1 montre laugmentation du pourcentage de femmes en aménorrhée au cours de létude de 12 mois. La figure 2 présente le pourcentage de femmes ayant présenté un spotting uniquement, des saignements uniquement, ou à la fois des saignements et un spotting au cours de la même période. Outre laménorrhée, laltération du cycle menstruel sest également caractérisée par des saignements intermenstruels, une ménorragie ou une métrorragie. Si les saignements anormaux associés à lAMPR en injection sous-cutanée persistent ou sils sont sévères, des examens appropriés doivent être effectués et un traitement doit être instauré.
Figure 1. Pourcentage de femmes traitées par AMPR en injection sous-cutanée ayant présenté une aménorrhée dans les études évaluant leffet contraceptif, par mois de 30 jours (population ITT, N = 2 053)
Figure 2. Pourcentage de femmes traitées par AMPR en injection sous-cutanée ayant présenté des saignements et/ou un spotting dans les études évaluant leffet contraceptif, par mois de 30 jours (population ITT, N = 2 053)
Risques de cancer :
La surveillance cas-témoins à long terme des utilisatrices dAMPR IM à 150 mg a montré labsence daugmentation globale du risque de cancer ovarien, hépatique ou cervical, et un effet protecteur prolongé sur la réduction du risque de cancer de lendomètre dans cette population.
Le cancer du sein est rare chez les femmes de moins de 40 ans, quelles utilisent ou pas une contraception hormonale.
Les résultats de quelques études épidémiologiques suggèrent une petite différence de risque de contracter la maladie chez les utilisatrices actuelles et récentes, comparativement aux femmes non utilisatrices. Le risque supplémentaire chez les utilisatrices actuelles et récentes dAMPR est faible par rapport au risque global de cancer du sein, en particulier chez les femmes jeunes (voir ci-dessous), et napparaît pas 10 ans après la dernière prise. La durée dutilisation ne semble pas être importante.
Nombre possible de cas supplémentaires de cancer du sein diagnostiqués jusquà 10 ans après larrêt des progestatifs injectables*
|
Age lors de la dernière utilisation dAMPR |
Nombre de cas pour 10 000 femmes naïves dutilisation |
Cas supplémentaires possibles pour 10 000 utilisatrices dAMPR |
|
20 |
Moins de 1 |
Bien moins que 1 |
|
30 |
44 |
2-3 |
|
40 |
160 |
10 |
*sur la base dune utilisation pendant 5 ans
Affections thromboemboliques
Bien quaucun lien de causalité nait été établi entre lAMP et la survenue daffections thrombotiques ou thromboemboliques, toute patiente qui développe ce type dévénement, comme une embolie pulmonaire, une maladie cérébrovasculaire ou une thrombose rétinienne ou une thrombose veineuse profonde, pendant le traitement par SAYANAPRESS, ne doit plus prendre le médicament. Les femmes ayant des antécédents daffections thromboemboliques nont pas été étudiées dans les essais cliniques et aucune information appuyant la sécurité demploi de SAYANAPRESS nest disponible pour cette population.
Anaphylaxie et réaction anaphylactoïde
Lors dune réaction anaphylactique, un traitement adapté sera instauré. Les réactions anaphylactiques graves exigent un traitement médical en urgence.
Affections oculaires
En cas de perte soudaine de la vue, partielle ou complète, ou en cas dapparition soudaine dune exophtalmie, dune diplopie ou dune migraine, le médicament ne doit pas être réadministré tant quun examen na pas été pratiqué. Si lexamen révèle un dème papillaire ou des lésions vasculaires de la rétine, le médicament ne doit pas être réadministré.
Précautions demploi
Variations de poids
Les variations de poids sont fréquentes mais imprévisibles. Dans les études de phase 3, le poids corporel a été suivi pendant 12 mois. La moitié (50 %) des femmes présentait une fluctuation de leur poids corporel initial de lordre de 2,2 kg. 12 % des femmes ont perdu plus de 2,2 kg et 38 % des femmes ont pris plus de 2,3 kg.
Rétention hydrique
Des données indiquent que les progestatifs peuvent entraîner un certain degré de rétention liquidienne ; par conséquent, le médicament devra être administré avec précaution chez toute patiente présentant une affection médicale préexistante susceptible dêtre affectée par la rétention hydrique.
Retour de lovulation
Après une dose unique dAMPR en injection sous-cutanée, le taux cumulé de retour de lovulation, mesuré par le taux plasmatique de la progestérone, était de 97,4 % (38 patientes sur 39) dans lannée suivant ladministration. Après la fenêtre thérapeutique de 14 semaines, le retour à lovulation le plus précoce est survenu dans un délai dune semaine, et le délai médian était de 30 semaines. Les femmes doivent être avisées du risque potentiel de retour retardé de lovulation après lutilisation de cette méthode, quelle que soit la durée dutilisation. Toutefois, il est établi que laménorrhée et/ou les irrégularités menstruelles qui suivent larrêt de la contraception hormonale peuvent être dues à un trouble sous-jacent associé à des irrégularités menstruelles, notamment à un syndrome des ovaires polykystiques.
Affections psychiatriques
Les patientes ayant déjà été traitées pour une dépression doivent faire lobjet dune surveillance rigoureuse pendant la prise de SAYANAPRESS.
Protection contre les maladies sexuellement transmissibles
Les patientes doivent être avisées du fait que SAYANAPRESS ne protège pas de linfection par le VIH (SIDA) ou des autres maladies sexuellement transmissibles.
Glucides/Métabolisme
Certaines patientes recevant des progestatifs peuvent présenter une diminution de la tolérance au glucose. Les patientes diabétiques doivent faire lobjet dune surveillance rigoureuse pendant la prise de ce type de traitement.
Fonction hépatique
Si une femme développe une jaunisse pendant la prise de SAYANAPRESS, il faut envisager de ne pas poursuivre le traitement (voir rubrique 4.3).
Hypertension et troubles lipidiques
Des données limitées suggèrent une légère augmentation du risque dévénements cardiovasculaires chez les femmes présentant une hypertension ou des anomalies lipidiques qui ont utilisé des progestatifs seuls sous forme injectable. Si une hypertension apparaît pendant le traitement par SAYANAPRESS et/ou si laugmentation de lhypertension ne peut être correctement contrôlée avec des antihypertenseurs, le traitement par SAYANAPRESS doit être interrompu. Les autres facteurs de risque daffections thrombotiques artérielles comprennent : lhypertension, le tabagisme, lâge, les anomalies lipidiques, la migraine, lobésité, les antécédents familiaux, les valvulopathies cardiaques, la fibrillation auriculaire.
SAYANAPRESS doit être utilisé avec prudence chez les patientes présentant au moins lun de ces facteurs de risque.
Autres affections
Les affections suivantes ont été rapportées pendant la grossesse et pendant lutilisation dhormones sexuelles stéroïdiennes, mais aucun lien na été établi avec lutilisation de progestatifs : une jaunisse et/ou un prurit associé à une cholestase, la formation de calculs biliaires, une porphyrie, un lupus érythémateux disséminé, un syndrome hémolytique et urémique, une chorée de Sydenham, un herpès gravidique, une perte auditive liée à une otosclérose.
En présence de lun(e) de ces affections/facteurs de risque, les bénéfices de lutilisation de SAYANAPRESS doivent être évalués individuellement en fonction des risques éventuels pour chaque femme et faire lobjet dune discussion avec la patiente avant quelle décide de prendre le médicament. En cas daggravation, dexacerbation ou dapparition de lun(e) de ces affections ou facteurs de risque, la patiente doit contacter son médecin. Le médecin devra ensuite déterminer si lutilisation de SAYANAPRESS doit être interrompue.
Analyses de laboratoire
Le pathologiste doit être avisé du traitement par un progestatif lorsque les échantillons concernés lui sont transmis. Le médecin doit être informé que certains tests endocriniens et de la fonction hépatique, ainsi que des paramètres sanguins, peuvent être affectés lors dun traitement par un progestatif.
a) Diminution des stéroïdes plasmatiques/urinaires (par exemple, progestérone, stradiol, prégnanediol, testostérone, cortisol)
b) Diminution des taux plasmatiques et urinaires de gonadotrophine (par exemple, LH, FSH)
c) Diminution des concentrations de globuline de liaison aux hormones sexuelles (SHBG).
Informations importantes au sujet des excipients
Ce produit contenant du parahydroxbenzoate de méthyle (E218) et du parahydroxbenzoate de propyle (E216), il peut provoquer des réactions allergiques (éventuellement retardées) et exceptionnellement, un bronchospasme. Ce médicament contient moins d1 mmol de sodium (23 mg) pour 104 mg/ 0,65 ml, cest à dire « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude dinteraction na été réalisée avec SAYANAPRESS.
De rares cas dinteractions avec dautres traitements médicaux (notamment des anticoagulants oraux) ont été rapportés, mais le lien de causalité na pas été établi. La possibilité dinteractions doit être prise en compte chez les patientes recevant des traitements concomitants.
In vitro, lAMP est métabolisé essentiellement par hydroxylation via le CYP3A4. Aucune étude dinteractions médicamenteuses spécifiques évaluant les effets cliniques avec des inducteurs ou des inhibiteurs du CYP3A4 sur lAMP na été réalisée ; par conséquent, les effets cliniques des inducteurs ou des inhibiteurs du CYP3A4 ne sont pas connus.
Une étude a révélé que les nourrissons issus de grossesses accidentelles survenues 1 à 2 mois après linjection IM dacétate de médroxyprogestérone à 150 mg présentaient un risque accru de faible poids de naissance, ce qui a été associé à un risque majoré de mort néonatale. Cependant, le risque global dun tel phénomène est très faible car les grossesses pendant le traitement par lacétate de médroxyprogestérone à 150 mg en injection IM sont peu fréquentes.
Les enfants exposés à lAMP in utero et suivis jusquà ladolescence nont présenté aucun signe deffet indésirable sur leur santé, notamment leur développement physique, intellectuel, sexuel ou social.
Allaitement
De faibles quantités détectables de médicament ont été retrouvées dans le lait de mères recevant de lAMP. Chez les femmes qui allaitent et qui reçoivent des injections IM dacétate de médroxyprogestérone à 150 mg, la composition, la qualité et la quantité de lait ne sont pas affectées. Les effets sur le développement et sur le comportement ont été étudiés chez les nouveau-nés et les nourrissons exposés à lAMP, par le biais du lait maternel, jusquà la puberté. Aucun effet indésirable na été constaté. Cependant, en raison de données limitées sur les effets de lAMP chez les nourrissons allaités de moins de 6 semaines, SAYANAPRESS doit être administré au moins 6 semaines après laccouchement, lorsque le système enzymatique du nourrisson est mieux développé.
Fertilité
SAYANAPRESS est indiqué pour la prévention dune grossesse.
Les femmes peuvent présenter un retour de la fertilité (conception) retardé après larrêt de SAYANAPRESS (voir rubrique 4.4).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
SAYANAPRESS na aucun effet sur laptitude à conduire des véhicules et à utiliser des machines.
Evénements observés lors des essais cliniques :
Dans trois vastes essais cliniques ayant inclus 1 980 femmes traitées par lAMPR en injection
sous-cutanée pendant une durée allant jusquà 1 an, les événements indésirables suivants ont été rapportés comme étant liés au médicament. Les effets indésirables sont répertoriés selon les catégories suivantes :
Très fréquent (³ 1/10)
Fréquent (³ 1/100 à < 1/10)
Peu fréquent (³ 1/1 000 à < 1/100)
Rare (³ 1/10 000 à < 1/1 000)
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
Evénements observés lors de la surveillance post-commercialisation :
Par ailleurs, les événements indésirables cliniquement significatifs extraits des données post-commercialisation de lAMPR injectable (IM ou SC) sont également inclus dans la liste ci-dessous :
|
Classe de systèmes dorganes |
Très fréquent |
Fréquent
|
Peu fréquent
|
Rare |
|
|
Infections et infestations |
|
Vaginite |
|
|
|
|
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes) |
|
|
|
Cancer du sein (voir rubrique 4.4) |
|
|
Affections du système immunitaire |
|
|
|
|
Réactions dhypersensibilité (par exemple, anaphylaxie et réactions anaphylactoïdes, angioedème (voir rubrique 4.4)) |
|
Troubles du métabolisme et de la nutrition |
Prise de poids, perte de poids (voir rubrique 4.4) |
|
Rétention hydrique (voir rubrique 4.4), augmentation de lappétit, diminution de lappétit |
|
|
|
Affections psychiatriques |
|
Dépression, anorgasmie, anxiété, trouble émotionnel, trouble affectif, diminution de la libido, irritabilité |
Insomnie, nervosité |
|
|
|
Affections du système nerveux |
|
Sensation vertigineuse, céphalée |
Migraine, somnolence |
|
Convulsions
|
|
Affections de loreille et du labyrinthe |
|
|
Vertige |
|
|
|
Affections cardiaques |
|
|
Tachycardie |
|
|
|
Affections vasculaires |
|
|
Embolie pulmonaire, thrombophlébite, hypertension (voir rubrique 4.4), varice, bouffées de chaleur |
|
Affections thromboemboliques (voir rubrique 4.4) |
|
Affections gastro-intestinales |
|
Douleur abdominale, nausées
|
Distension abdominale |
|
|
|
Affections hépatobiliaires |
|
|
Taux denzyme hépatique anormal
|
|
Jaunisse, perturbation de la fonction hépatique (voir rubrique 4.4) |
|
Affections de la peau et du tissu sous-cutané |
|
Acné |
Chloasma, dermatite, ecchymose, rash, alopécie, hirsutisme, prurit, urticaire |
|
Strie cutanée |
|
Affections musculo-squelettiques et systémiques |
|
Dorsalgie, douleur dans les membres |
Perte de la densité minérale osseuse (voir rubrique 4.4), arthralgie, crampes musculaires |
|
Ostéoporose, incluant des fractures ostéoporotiques |
|
Affections des organes de reproduction et du sein |
|
Aménorrhée, douleur/ hypersensibilité mammaire, dysménorrhée, métrorragie, ménométrorragie, ménorragie (voir rubrique 4.4) |
Kyste de lovaire, hémorragie utérine (irrégulière, augmentée, diminuée), pertes vaginales, sécheresse vulvo-vaginale, accroissement mammaire, dyspareunie, galactorrhée, douleur pelvienne, syndrome prémenstruel |
|
|
|
Troubles généraux et anomalies au site dadministration |
|
Réactions au site dinjection (telles quune douleur au point dinjection, une sensibilité au niveau du site dinjection, un nodule au site dinjection, une atrophie (persistante) au site dinjection et une lipoatrophie au site dinjection), fatigue |
Asthénie |
Fièvre |
|
|
Investigations |
|
Frottis cervical anormal
|
Tolérance diminuée au glucose (voir rubrique 4.4) |
|
|
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Aucune action nest requise outre linterruption du traitement.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : progestatifs, code ATC : G03AC06.
LAMP est un analogue de la 17 a‑hydroxyprogestérone avec des effets anti-strogéniques, antiandrogéniques et antigonadotrophiques.
LAMPR en injection sous-cutanée inhibe la sécrétion des gonadotrophines ce qui, à son tour, prévient la maturation folliculaire et lovulation. Le principal mécanisme de suppression de lovulation entraîne également un amincissement de lendomètre, et ces actions produisent son effet contraceptif.
Modifications de la DMO chez les Femmes Adultes
Une étude comparant les modifications de la DMO chez des femmes utilisant de lAMPR en injection sous-cutanée et chez des femmes recevant de lacétate de médroxyprogestérone injectable (150 mg IM) na pas montré de différences significatives de perte de DMO entre les deux groupes après deux ans de traitement. Les pourcentages moyens de modification de la DMO dans le groupe AMPR en injection sous-cutanée sont indiqués dans le tableau 1.
Tableau 1. Modification moyenne (%) de la DMO par rapport aux valeurs initiales chez des femmes prenant SAYANAPRESS, par site squelettique
|
Durée du traitement |
Rachis lombaire |
Hanche totale |
Col du fémur |
|||
|
N |
Modification (%) (IC à 95 %) |
N |
Modification (%) (IC à 95 %) |
N |
Modification (%) (IC à 95 %) |
|
|
1 an |
166 |
-2,7 (-3,1 à -2,3) |
166 |
-1,7 (-2,1 à -1,3) |
166 |
-1,9 (-2,5 à -1,4) |
|
2 ans |
106 |
- 4,1 (-4,6 à -3,5) |
106 |
-3,5 (-4,2 à -2,7) |
106 |
-3,5 (-4,3 à -2,6) |
Une autre étude clinique contrôlée menée chez des femmes adultes prenant de lacétate de médroxyprogestérone injectable (150 mg IM) pendant une durée maximale de 5 ans a montré des diminutions moyennes de la DMO de lordre de 5-6 % au niveau du rachis et de la hanche, et aucun changement significatif de la DMO dans le groupe témoin. La diminution de la DMO était plus marquée durant les deux premières années dutilisation, avec des diminutions plus faibles au cours des années suivantes. Des modifications moyennes de la DMO de lordre de -2,86 %, -4,11 %, -4,89 %, -4,93 % et -5,38 % ont été observées au niveau du rachis lombaire après 1, 2, 3, 4 et 5 ans, respectivement. Les diminutions moyennes de la DMO au niveau de la hanche totale et du col du fémur étaient similaires. Voir le tableau 2 ci-dessous pour plus de détails.
Après larrêt des injections dacétate de médroxyprogestérone (150 mg IM), la DMO a augmenté vers les valeurs initiales au cours de la période post-traitement. Une durée de traitement plus longue a été associée à une récupération plus lente de la DMO.
Tableau 2. Modification moyenne (%) de la DMO par site squelettique par rapport aux valeurs initiales chez des femmes adultes, de la cohorte traitées pendant 5 ans par lacétate de médroxyprogestérone à 150 mg IM et après 2 ans post-traitement ou après 7 ans dobservation chez des femmes du groupe témoin
|
Durée dinclusion dans létude |
Rachis |
Hanche totale |
Col du fémur |
|||
|
|
AMP |
Groupe témoin |
AMP |
Groupe témoin |
AMP |
Groupe témoin |
|
5 ans* |
n = 33 -5,38 % |
n = 105 0,43 % |
n = 21 -5,16 % |
n = 65 0,19 % |
n = 34 -6,12 % |
n = 106 -0,27 % |
|
7 ans** |
n = 12 -3,13 % |
n = 60 0,53 % |
n = 7 -1,34 % |
n = 39 0,94 % |
n = 13 -5,38 % |
n = 63 -0,11 % |
*Le groupe de traitement se composait de femmes ayant reçu de lacétate de médroxyprogestérone en injection (150 mg IM) pendant 5 ans, et le groupe témoin se composait de femmes nayant pas utilisé de contraception hormonale au cours de cette période.
**Le groupe de traitement se composait de femmes ayant reçu de lacétate de médroxyprogestérone en injection (150 mg IM) pendant 5 ans puis suivies pendant une période maximale de 2 ans post-traitement, et le groupe témoin se composait de femmes nayant pas utilisé de contraception hormonale pendant 7 ans.
Modifications de la DMO chez les Adolescentes (12-18 ans)
Les résultats dune étude clinique non randomisée en ouvert portant sur lutilisation de lacétate de médroxyprogestérone en injection (150 mg IM toutes les 12 semaines pendant une période de 240 semaines [4,6 ans] maximum, suivie de mesures post-traitement) chez des adolescentes (12-18 ans) ont également montré que lutilisation dacétate de médroxyprogestérone IM était associée à une diminution significative de la DMO par rapport aux valeurs initiales. Chez les patientes qui recevaient ≥ 4 injections/période de 60 semaines, la diminution moyenne de la DMO au niveau du rachis lombaire était de -2,1 % au bout de 240 semaines (4,6 ans) ; les diminutions moyennes de la DMO au niveau de la hanche totale et du col du fémur étaient de -6,4 % et -5,4 %, respectivement. Le suivi post-traitement a montré que, sur la base des valeurs moyennes, la DMO au niveau du rachis lombaire retrouvait les valeurs initiales environ 1 an après larrêt du traitement et que la DMO au niveau de la hanche retrouvait les valeurs initiales environ 3 ans après larrêt du traitement. Toutefois, il est important de noter quun grand nombre de sujets sont sortis détude, et que ces résultats se basent donc sur un petit nombre de sujets (n = 71 à 60 semaines et n = 25 à 240 semaines après larrêt du traitement). En revanche, une cohorte non comparable de sujets non traités et non appariés, présentant des paramètres osseux initiaux différents de ceux des utilisatrices dAMPR, a montré des augmentations moyennes de la DMO à 240 semaines de lordre de 6,4 %, 1,7 % et 1,9 % au niveau du rachis lombaire, de la hanche totale et du col du fémur, respectivement.
5.2. Propriétés pharmacocinétiques
|
Tableau 1. Paramètres pharmacocinétiques de lAMP après une injection SC unique de SAYANAPRESS chez des femmes en bonne santé (n = 42) |
||||||
|
|
Cmax (ng/ml) |
Tmax (jour) |
C91 (min) (ng/ml) |
ASC0-91 (ng·jour/ml) |
ASC0-¥ (ng·jour/ml) |
t½ (jour) |
|
Moyenne |
1,56 |
8,8 |
0,402 |
66,98 |
92,84 |
43 |
|
Min |
0,53 |
2,0 |
0,133 |
20,63 |
31,36 |
16 |
|
Max |
3,08 |
80,0 |
0,733 |
139,79 |
162,29 |
114 |
|
Cmax = pic sérique ; Tmax = temps quand Cmax est observé, ASC0-91 = aire sous la courbe concentration-temps sur 91 jours ; t½ = demi-vie terminale ; 1 nanogramme = 103 picogrammes.
|
||||||
Caractéristiques générales
Absorption
Labsorption de lAMP du site dinjection SC pour atteindre les niveaux thérapeutiques est relativement rapide. Le Tmax moyen était atteint une semaine environ après linjection. Les concentrations maximales dAMP (Cmax) sont généralement comprises entre 0,5 et 3,0 ng/ml avec une Cmax moyenne de 1,5 ng/ml après une injection SC unique.
Effet du site dinjection
LAMPR était administré par injection sous cutanée au niveau de la partie antérieure de la cuisse ou de labdomen afin den évaluer les effets sur le profil concentration-temps de lAMP. Les concentrations minimales dAMP (Cmin ; Jour 91) étaient similaires pour les deux sites dinjection, ce qui suggère que le site dinjection ne nuit pas à lefficacité contraceptive.
Distribution
La liaison de lAMP aux protéines plasmatiques avoisine 86 %. LAMP se lie essentiellement à lalbumine sérique ; il ne se fixe pas à la SHBG.
Biotransformation
LAMP est fortement métabolisé dans le foie par les enzymes P450. Son métabolisme porte essentiellement sur une réduction de lanneau A et/ou de la chaîne latérale, une perte du groupe acétyle, une hydroxylation en position 2, 6 et 21 ou une combinaison de ces positions, donnant lieu à plus de 10 métabolites.
Élimination
Les concentrations résiduelles dAMP au terme de lintervalle dadministration (3 mois) dAMPR en injection sous-cutanée sont généralement inférieures à 0,5 ng/ml, ce qui est en accord avec sa demi-vie terminale apparente denviron 40 jours après une administration SC. La plupart des métabolites de lAMP sont excrétés dans les urines sous forme de glucuroconjugués, avec seulement de faibles quantités excrétées sous forme de sulfates.
Linéarité/non-linéarité
Les données relatives à lutilisation de doses uniques nont pas révélé de relation non linéaire pour des doses de 50 à 150 mg après ladministration SC. La relation entre lASC ou la Cmin et la dose SC dAMP sest révélée linéaire. La Cmax moyenne na pas varié de manière substantielle avec laugmentation de la dose.
Race
Il ny a pas eu de différence apparente de profil pharmacocinétique et/ou pharmacodynamique de lAMP après ladministration SC dAMPR aux femmes de toutes les origines ethniques étudiées. Le profil pharmacocinétique/pharmacodynamique de lAMP a été évalué chez des femmes asiatiques dans le cadre dune étude séparée.
Effet du poids corporel
Aucun ajustement posologique de SAYANAPRESS nest nécessaire en fonction du poids corporel. Leffet du poids corporel sur les paramètres pharmacocinétiques de lAMP a été évalué dans un sous-ensemble de femmes (n = 42, indice de masse corporelle [IMC] compris entre 18,2 et 46,0 kg/m2). Les valeurs dASC0-91 pour lAMP étaient de 68,5 ; 74,8 et 61,8 ng - jour/ml chez les femmes appartenant aux catégories dIMC £ 25 kg/m2, > 25 à ≤ 30 kg/m2, et > 30 kg/m2, respectivement. La Cmax moyenne de lAMP était de 1,65 ng/ml chez les femmes ayant un IMC £ 25 kg/m2, de 1,76 ng/ml chez les femmes ayant un IMC compris entre > 25 et ≤ 30 kg/m2, et de 1,40 ng/ml chez les femmes ayant un IMC > 30 kg/m2. La fourchette des valeurs des concentrations minimales (Cmin) et des demi-vies de lAMP étaient comparables pour les 3 groupes dIMC.
Relation(s) pharmacocinétique/pharmacodynamique
Dun point de vue pharmacodynamique, la durée de la suppression de lovulation dépend du maintien des concentrations thérapeutiques de lAMP tout au long de lintervalle dadministration de 13 semaines.
5.3. Données de sécurité préclinique
Parahydroxybenzoate de méthyle (E218)
Parahydroxybenzoate de propyle (E216)
Chlorure de sodium
Polysorbate 80
Phosphate monosodique monohydraté
Phosphate disodique dodécahydraté
Méthionine
Povidone
Acide chlorhydrique et/ou hydroxyde de sodium pour lajustement du pH
Eau pour préparations injectables
Après ouverture : A utiliser immédiatement. Eliminer toute portion inutilisée.
6.4. Précautions particulières de conservation
Ne pas mettre au réfrigérateur. Ne pas congeler.
6.5. Nature et contenu de l'emballage extérieur
6.6. Précautions particulières délimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Instructions dutilisation et de manipulation
|
Préparatifs |
|
|
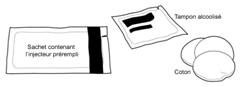
· Vérifier que le médicament est à température ambiante. · Sassurer que les éléments suivants sont disponibles : o Un sachet en aluminium fermé contenant SAYANAPRESS dans linjecteur prérempli o Un tampon alcoolisé o Un morceau de coton propre
(Le tampon alcoolisé et le coton ne sont pas fournis avec SAYANAPRESS.)
|
|
|
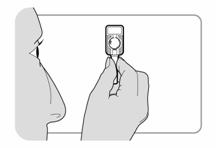
Etape 1 : Choix et préparation de la zone dinjection |
|
|
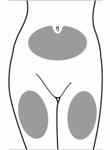
· Choisir une zone adaptée pour linjection sous-cutanée, à savoir labdomen ou lavant du haut de la cuisse. Eviter les zones osseuses et le nombril. · Utiliser un tampon alcoolisé pour nettoyer la peau de la zone dinjection choisie. Laisser la peau sécher.
|
|
|
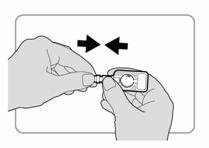
Etape 2 : Préparation de linjecteur |
|
|
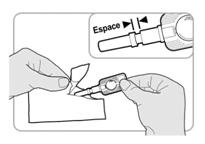
· Au moment dadministrer linjection, déchirer soigneusement le sachet en aluminium et sortir linjecteur. Ne pas retirer le protecteur daiguille à ce stade. · Vérifier linjecteur comme suit : o Le protecteur daiguille doit être dans la position indiquée sur lillustration. Il doit y avoir un espace entre lextrémité du protecteur daiguille et le porte-aiguille. o En labsence despace, jeter linjecteur et en utiliser un autre. o Si le protecteur daiguille sest désolidarisé de laiguille, ou sil est manquant, jeter linjecteur et en utiliser un autre.
|
|
|
Etape 3 : Mélange du médicament |
|
|
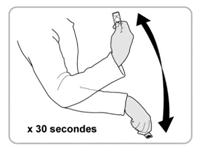
· Bien tenir linjecteur par le porte-aiguille (voir la Figure 1 pour connaître lemplacement du porte-aiguille) · Agiter vigoureusement linjecteur pendant 30 secondes pour bien mélanger le médicament. Ne pas le plier. · En cas de délai entre le mélange du médicament et le passage aux étapes suivantes, renouveler la procédure de mélange ci-dessus. |
|
|
· Vérifier linjecteur. Le liquide à lintérieur doit être de couleur blanche à blanc cassé et uniforme. Il ne doit présenter aucune fuite. · En cas de problème, jeter linjecteur et en utiliser un autre. |
|
|
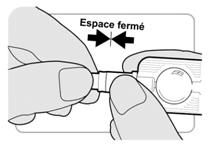
Etape 4 : Activation de linjecteur |
|
|
· Bien tenir linjecteur par le porte-aiguille dune main. · Tenir le protecteur daiguille avec lautre main. Il y a un espace entre le porte-aiguille et lextrémité du protecteur daiguille. |
|
|
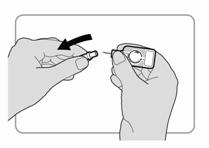
· Pousser le protecteur daiguille vers le porte-aiguille. Continuer de presser fermement jusquà ce quil ny ait plus despace entre le protecteur daiguille et le porte-aiguille. Linjecteur est à présent activé. |
|
|
· Continuer de bien tenir linjecteur par le porte-aiguille. · Tirer sur le protecteur daiguille pour le retirer de laiguille.
|
|
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
23-25 AVENUE DU DR LANNELONGUE
75014 PARIS
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 300 078 6 0 : injecteur prérempli contenant 0,65 ml de suspension injectable. Boite de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[A compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[A compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I