RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 15/01/2016
![]() Ce médicament fait lobjet dune surveillance supplémentaire qui permettra lidentification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait lobjet dune surveillance supplémentaire qui permettra lidentification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
ACARIZAX 12 SQ-HDM, lyophilisat oral
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour la liste complète des excipients, voir rubrique 6.1.
* SQ-HDM est lunité de dose pour ACARIZAX. SQ est une méthode de standardisation de la puissance biologique, de la teneur en allergènes majeurs et de la complexité de lextrait allergénique. HDM est labréviation de house dust mite (acarien de la poussière de maison)
Lyophilisat oral blanc à blanc cassé en creux.
4.1. Indications thérapeutiques
· une rhinite allergique persistante modérée à sévère aux acariens insuffisamment contrôlée par les traitements symptomatiques
et/ou
· un asthme allergique aux acariens insuffisamment contrôlé par les corticostéroïdes inhalés et associé à une rhinite allergique légère à sévère aux acariens. Lasthme du patient doit être soigneusement évalué avant linstauration du traitement (voir rubrique 4.3).
4.2. Posologie et mode d'administration
La posologie recommandée chez ladulte est d1 lyophilisat oral (12 SQ-HDM) par jour.
Lapparition de leffet clinique est attendue 8 à 14 semaines après linstauration du traitement. Les recommandations thérapeutiques internationales préconisent une durée d'immunothérapie allergénique d'environ 3 ans pour modifier lévolution de la maladie. Les données d'efficacité avec ACARIZAX sont disponibles sur une période de 18 mois de traitement chez l'adulte ; aucune donnée nest disponible sur une période de 3 ans de traitement (voir rubrique 5.1). S'il nest pas observé d'amélioration pendant la première année de traitement par ACARIZAX, la poursuite du traitement nest pas justifiée.
Population pédiatrique
Lexpérience clinique dune immunothérapie par ACARIZAX n'est pas disponible chez lenfant de moins de 18 ans. ACARIZAX nest pas indiqué chez l'enfant de moins de 18 ans. Les données actuellement disponibles chez lenfant sont décrites dans la rubrique 5.1.
Patients âgés
Lexpérience clinique dune immunothérapie par ACARIZAX n'est pas disponible chez le sujet de plus de 65 ans. ACARIZAX nest pas indiqué chez le sujet de plus de 65 ans (voir rubrique 5.1).
Mode dadministration
Le traitement par ACARIZAX doit être instauré par des médecins expérimentés dans le traitement des allergies.
La première prise de lyophilisat oral devra être réalisée sous surveillance médicale pendant au moins 30 minutes afin d'évaluer et traiter les éventuels effets indésirables d'apparition immédiate,
ACARIZAX est un lyophilisat oral. La plaquette thermoformée sera ouverte avec des doigts bien secs pour libérer le lyophilisat oral. Immédiatement après sa libération de la plaquette thermoformée, le lyophilisat oral doit être placé sous la langue où il se dissout. Ne pas déglutir pendant environ 1 minute. Ne pas absorber daliment ni de boisson dans les 5 minutes qui suivent la prise du médicament.
Si le traitement par ACARIZAX est interrompu pendant une durée allant jusquà 7 jours, le patient peut reprendre le traitement de lui-même. Si le traitement est interrompu pendant plus de 7 jours, un avis médical est recommandé pour la reprise éventuelle du traitement.
· hypersensibilité à lun des excipients (pour la liste complète des excipients, voir rubrique 6.1).
· VEMS < 70 % de la valeur théorique (après un traitement médicamenteux adapté) lors de l'initiation du traitement.
· exacerbation sévère dasthme au cours des 3 derniers mois.
· infection aiguë des voies respiratoires chez les sujets asthmatiques; la mise en route du traitement par ACARIZAX doit être différée jusquà la guérison de linfection respiratoire
· maladies auto-immunes évolutives ou mal contrôlées, déficits immunitaires, immunodépression ou maladies néoplasiques malignes évolutives.
· inflammation buccale aiguë sévère ou plaies de la muqueuse buccale (voir rubrique 4.4).
4.4. Mises en garde spéciales et précautions d'emploi
Lasthme est un facteur de risque dans la survenue de réactions allergiques systémiques sévères.
Les patients seront informés du fait qu'ACARIZAX nest pas indiqué pour le traitement des exacerbations aiguës dasthme. Dans ce cas, un bronchodilatateur de courte durée d'action doit être utilisé. Un avis médical est requis en cas de perte d'efficacité des bronchodilatateurs ou d'augmentation de leur consommation par le patient.
Les patients doivent être informés de la nécessité de consulter immédiatement un médecin en cas d'aggravation aiguë de leur asthme.
ACARIZAX doit être initialement utilisé en complément du traitement antiasthmatique en cours, et non en remplacement de celui-ci.
Les traitements en cours pour le contrôle de l'asthme ne doivent pas être arrêtés brutalement après linstauration d'un traitement par ACARIZAX. La diminution des doses des médicaments administrés pour le contrôle de lasthme ne doit être envisagée que de façon progressive et sous contrôle médical selon les recommandations de prise en charge de lasthme.
Réactions allergiques systémiques sévères
Le traitement doit être interrompu et un avis médical immédiat est requis en cas de réaction allergique systémique sévère, d'exacerbation sévère dasthme, dangio-dème, de dysphagie, de dyspnée, de dysphonie, dhypotension ou de sensation de constriction pharyngée. Les symptômes annonciateurs d'une réaction systémique peuvent inclure des flushs (bouffées vasomotrices), un prurit, une sensation de chaleur, un malaise général et une agitation ou une anxiété.
L'adrénaline peut être nécessaire pour traiter des réactions allergiques systémiques sévères. Les traitements concomitants par antidépresseurs tricycliques, inhibiteurs de la monoamine oxydase (IMAO) et/ou inhibiteurs de la catéchol-O-méthyltransférase (COMT), peuvent potentialiser les effets de ladrénaline et mettre en jeu le pronostic vital. Les effets de ladrénaline peuvent être diminués chez les patients traités par bêtabloquants.
Les patients présentant une cardiopathie peuvent être exposés à un risque plus important en cas de réactions allergiques systémiques. Lexpérience clinique du traitement par ACARIZAX chez les patients présentant une cardiopathie est limitée. Ceci doit être pris en considération avant dinstaurer une immunothérapie allergénique.
Linstauration dun traitement par ACARIZAX chez les patients ayant déjà présenté une réaction allergique systémique au cours d'une immunothérapie aux acariens par voie sous-cutanée doit être envisagée avec précaution, en ayant à disposition les traitements nécessaires en cas de survenue de réactions indésirables. Lexpérience acquise depuis la commercialisation dun comprimé sublingual similaire destiné au traitement de l'allergie aux pollens de graminées montrent que le risque de réaction allergique sévère peut être plus important chez les patients ayant déjà présentés une réaction allergique systémique au cours dune immunothérapie aux pollens de graminées par voie sous-cutanée.
Inflammation de la muqueuse buccale
En cas de d'inflammation sévère de la muqueuse buccale (exemple : lichen plan buccal, ulcérations ou mycose), de plaies dans la bouche, ou de chirurgie buccopharyngée, y compris une extraction dentaire, ou la perte dune dent, linstauration du traitement par ACARIZAX sera différée, ou le traitement en cours temporairement interrompu, jusqu'à la cicatrisation.
Réactions allergiques locales
Un traitement par ACARIZAX expose le patient aux allergènes auxquels il est allergique. De ce fait, la survenue de réactions allergiques locales est attendue au cours du traitement. Ces réactions sont généralement légères ou modérées, mais des réactions oropharyngées plus sévères peuvent survenir. En cas d'apparition de réactions indésirables locales significatives induites par l'administration d'ACARIZAX, l'utilisation d'un antihistaminique doit être envisagée.
sophagite à éosinophiles
Des cas isolés dsophagite à éosinophiles ont été rapportés au cours d'un traitement par ACARIZAX. Un avis médical est requis en cas de symptômes gastro-sophagiens sévères ou persistants, tels que dysphagie ou dyspepsie.
Maladies auto-immunes en rémission
Les données disponibles concernant un traitement par immunothérapie allergénique chez des patients présentant des maladies auto-immunes en rémission, sont limitées. ACARIZAX doit ainsi être prescrit avec précaution chez ces patients.
Allergie alimentaire
ACARIZAX peut contenir des traces de protéines de poisson. Les données disponibles nont pas mis en évidence de risque accru de réactions allergiques chez les patients présentant une allergie au poisson.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude dinteraction na été réalisée chez lhomme, et il n'a pas été rapporté d'interaction médicamenteuse potentielle. Ladministration concomitante de traitements symptomatiques de lallergie peut augmenter la tolérance clinique de limmunothérapie allergénique, ce qui doit être pris en considération à larrêt de ces médicaments.
Aucune donnée clinique nest disponible concernant lutilisation dACARIZAX chez la femme enceinte. Les études réalisées chez lanimal nindiquent pas de risque accru pour le ftus. Le traitement par ACARIZAX ne doit pas être instauré au cours de la grossesse. Si une grossesse survient en cours de traitement, la décision de poursuite ou non de l'immunothérapie allergénique devra prendre en considération létat clinique de la patiente (incluant la fonction respiratoire) ainsi que ses antécédents de réactions apparues lors des prises précédentes dACARIZAX. En cas dasthme préexistant, une surveillance étroite est recommandée pendant la grossesse.
Aucune donnée clinique nest disponible concernant lutilisation dACARIZAX au cours de lallaitement. Il nest pas attendu deffets particuliers chez les enfants allaités.
Fertilité
Aucune donnée clinique nest disponible concernant leffet dACARIZAX sur la fertilité. Lors dune étude de toxicité en administrations réitérées chez la souris, aucun effet na été observé sur les organes de reproduction des animaux des deux sexes.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables attendus au cours du traitement par ACARIZAX sont la survenue de réactions allergiques locales légères à modérées au cours des premiers jours du traitement, disparaissant en 1 à 3 mois avec la poursuite du traitement (voir rubrique 4.4). Dans la majorité des cas, les réactions apparaissent toujours dans les 5 minutes suivant la prise dACARIZAX et disparaissent en quelques minutes ou plusieurs heures. Des réactions allergiques oropharyngées plus sévères peuvent apparaître (voir rubrique 4.4).
Des cas isolés daggravation aiguë sévère des symptômes dasthme ont été rapportés. Le traitement par ACARIZAX ne doit pas être instauré chez les patients ayant des facteurs de risque connus (voir rubrique 4.3).
Tableau des effets indésirables
Le tableau des effets indésirables mentionné ci-dessous est établi à partir des données issues d'études cliniques contrôlées contre placebo réalisées chez des adultes atteints de rhinite et/ou dasthme allergique aux acariens traités par ACARIZAX.
Les réactions indésirables sont regroupées selon leur fréquence de survenue et conformément à la classification MedDRA : très fréquent (≥ 1/10), fréquent (≥1/100 à <1/10), peu fréquent (≥1/1 000 à <1/100), rare (≥1/10 000 à <1/1 000), très rare (<1/10 000).
|
Classes organiques |
Fréquence |
Effets indésirables |
|
Infections et infestations |
Très fréquent |
Rhinopharyngite |
|
Fréquent |
Bronchite, laryngite, pharyngite, rhinite, sinusite |
|
|
Troubles du système nerveux |
Peu fréquent |
Sensation vertigineuse, dysgueusie |
|
Troubles oculaires |
Fréquent |
Prurit oculaire |
|
Affections de loreille et du labyrinthe |
Fréquent |
Prurit auriculaire |
|
Troubles respiratoires, thoraciques et médiastinaux |
Très fréquent |
Irritation de la gorge |
|
Fréquent |
Dysphonie, dyspnée, douleur oropharyngée, dème pharyngé |
|
|
Peu fréquent |
dème laryngé, congestion nasale, gêne nasale, sensation d'obstruction nasale, rhinorrhée, éternuement, sensation de constriction pharyngée |
|
|
Troubles gastro-intestinaux |
Très fréquent |
dème buccal, prurit oral |
|
Fréquent |
Douleur abdominale, diarrhée, sécheresse buccale, dysphagie, dyspepsie, glossodynie, dème labial, prurit labial, prurit lingual, nausées, gêne buccale, paresthésie buccale, stomatite, dème lingual |
|
|
Peu fréquent |
Glossite, ulcération buccale, irritation sophagienne, vésicules buccales, érythème de la muqueuse buccale, vomissements |
|
|
Troubles généraux et anomalies au site dadministration |
Fréquent |
Gêne thoracique |
|
Peu fréquent |
Fatigue, malaise, sensation de corps étranger |
|
|
Troubles cutanés et du tissu sous-cutané |
Peu fréquent |
Prurit |
Description de certaines réactions indésirables
En cas d'apparition d'effets indésirables significatifs liés à l'administration dACARIZAX, le recours à un traitement symptomatique de lallergie doit être envisagé.
Des cas de réactions allergiques systémiques ont été rapportés avec un comprimé sublingual similaire destiné au traitement de lallergie aux pollens de graminées et sont considérés comme un effet de classe. La première administration du lyophilisat oral doit donc être réalisée sous surveillance médicale (voir rubrique 4.2).
En cas daggravation aiguë des symptômes dasthme ou de réactions allergiques systémiques sévères, dangio-dème, de dysphagie, de dyspnée, de dysphonie, dhypotension ou de sensation de constriction pharyngée, un avis médical immédiat est requis. Le traitement doit être interrompu définitivement ou jusquà avis contraire du médecin.
Des cas isolés dsophagite à éosinophiles ont été rapportés (voir rubrique 4.4).
Population pédiatrique
ACARIZAX nest pas indiqué chez les enfants de moins de 18 ans (voir rubrique 4.2). Des données limitées sont disponibles chez des enfants de 5 à 17 ans. Aucune donnée n'est disponible chez les enfants de moins de 5 ans traités par ACARIZAX.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Si des doses supérieures à la dose quotidienne recommandée sont prises, le risque deffets indésirables augmente, y compris le risque de réactions allergiques systémiques ou de réactions allergiques locales sévères. En cas de réaction sévère telle quun angio-dème, une dysphagie, une dyspnée, une modification de la voix ou une sensation de constriction pharyngée un avis médical immédiat est requis. Un traitement symptomatique adapté est préconisé.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : extraits allergéniques, acariens, code ATC : V01AA03.
Mécanisme daction
ACARIZAX est une immunothérapie allergénique. Limmunothérapie allergénique consiste en ladministration répétée dallergènes à un individu allergique dans le but de modifier sa réponse immunitaire à ces allergènes.
Lactivité pharmacodynamique de l'immunothérapie allergénique a pour cible le système immunitaire mais le mécanisme d'action exact à l'origine de leffet clinique nest pas totalement connu. Les travaux ont mis en évidence une augmentation de la production d'IgG4 spécifiques anti-acariens induits par l'administration d'ACARIZAX et la production danticorps circulants pouvant entrer en compétition au niveau de la liaison des IgE avec les allergènes dacariens. Cet effet a été observé dès quatre semaines de traitement.
ACARIZAX agit sur la cause de lallergie respiratoire aux acariens, et son effet clinique au cours du traitement a été démontré au niveau des voies respiratoires supérieures et inférieures. La protection induite par ACARIZAX a entraîné une amélioration du contrôle des symptômes et de la qualité de vie, liés à un soulagement des symptômes, une diminution du recours aux autres médicaments et la réduction du risque d'exacerbations.
Efficacité clinique chez ladulte
Lefficacité dACARIZAX 12 SQ-HDM dans le traitement des allergies respiratoires aux acariens a été évaluée au cours de deux études cliniques randomisées, en double aveugle, contrôlées contre placebo, menées dans des populations différentes et utilisant des critères primaires différents. Deux tiers des patients inclus dans ces études étaient sensibilisés aux acariens et à d'autres allergènes. Les résultats cliniques n'ont pas été influencés par le fait que les patients soient mono ou polysensibilisés. Des données complémentaires provenant dune étude réalisée en chambre dexposition aux allergènes et dune étude menée à de plus faibles doses sont également présentées.
Rhinite allergique
Létude MERIT (MT-06)
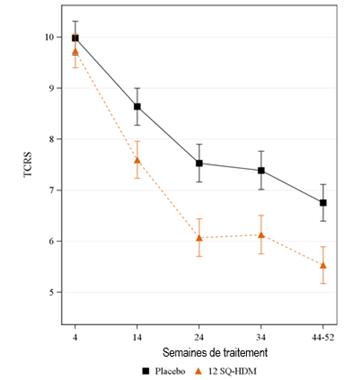
Létude MERIT a été menée chez 992 adultes présentant une rhinite allergique modérée à sévère aux acariens malgré un traitement symptomatique de la rhinite. Ces patients ont été randomisés pour recevoir une prise quotidienne de 12 SQ-HDM, de 6 SQ-HDM ou dun placebo pendant environ 1 an, et pouvaient recourir à un traitement symptomatique de la rhinite. Les patients ont été examinés par un spécialiste environ tous les deux mois durant la totalité de létude. Le critère primaire était le score total combiné quotidien moyen de rhinite (daily total combined rhinitis score, TCRS) évalué durant les 8 dernières semaines de traitement. Le TCRS était la somme du score des symptômes de rhinite et du score médicamenteux de la rhinite. Le score des symptômes de rhinite évaluait chacun des quatre symptômes nasaux (rhinorrhée, obstruction nasale, prurit nasal, éternuements) quotidiennement sur une échelle de 0 à 3 (symptômes absents, légers, modérés ou sévères) ; échelle de score allant de 0 à 12. Le score médicamenteux de la rhinite était la somme du score de la prise dun corticostéroïde nasal (2 points par bouffée, maximum 4 bouffées par jour) et du score de la prise dun antihistaminique oral (4 points par comprimé, maximum 1 comprimé par jour) ; échelle de score allant de 0 à 12. Léchelle de score du TCRS allait ainsi de 0 à 24. Les principaux critères secondaires prédéfinis étaient le score total combiné de rhinoconjonctivite et de qualité de vie liée à la rhinoconjonctivite (rhinoconjunctivitis quality of life, RQLQ). Des analyses des jours avec exacerbation de rhinite ont été également réalisées a posteriori pour illustrer davantage la pertinence clinique des résultats. Une exacerbation de rhinite était définie comme un jour où les symptômes étaient revenus au niveau élevé requis pour linclusion des patients dans létude : score de symptômes de rhinite dau moins 6, ou dau moins 5 en présence d'un symptôme côté comme sévère.
|
|
Etude MERIT : évolution du score total combiné de rhinite au cours du temps TCRS : score total combiné de rhinite (score des symptômes + score médicamenteux). Le critère primaire était le TCRS quotidien moyen au cours des 8 dernières semaines de traitement environ (semaines 44 à 52 environ). Moyennes ajustées du TCRS moyen au cours du temps. Les lignes verticales indiquent lerreur standard pour ces moyennes ajustées. Les intervalles ne se chevauchant pas indiquent une différence statistiquement significative. |
Résultats de l'étude MERIT |
12 SQ-HDM |
Placebo |
Efficacité |
|
|||
|
Critère primaire |
N |
Score |
N |
Score |
Différence absolue c |
Différence relative d |
Valeur de p |
|
Score total combiné de rhinite |
|||||||
|
FAS-MI a (moyenne ajustée) |
318 |
5,71 |
338 |
6,81 |
1,09 [0,35 ; 1,84] |
- |
0,004 |
|
FAS b (moyenne ajustée) |
284 |
5,53 |
298 |
6,76 |
1,22 [0,49 ; 1,96] |
18 % |
0,001 |
|
FAS b (médiane) |
284 |
5,88 |
298 |
7,54 |
1,66 |
22 % |
- |
|
Principaux critères secondaires prédéfinis |
N |
Score |
N |
Score |
Différence absolue c |
Différence relative d |
Valeur de p |
|
Score de symptômes de rhinite |
|||||||
|
FAS b (moyenne ajustée) |
284 |
2,76 |
298 |
3,30 |
0,54 [0,18 ; 0,89] |
16 % |
0,003 |
|
FAS b (médiane) |
284 |
2,98 |
298 |
3,98 |
1,00 |
25 % |
- |
|
Score médicamenteux de la rhinite |
|||||||
|
FAS b (moyenne ajustée) |
284 |
2,22 |
298 |
2,83 |
0,60 [0,08 ; 1,13] |
21 % |
0,024 |
|
FAS b (médiane) |
284 |
2,83 |
298 |
4,00 |
1,17 |
29 % |
- |
|
Score total combiné de rhinoconjonctivite |
|||||||
|
FAS b (moyenne ajustée) |
241 |
7,91 |
257 |
9,12 |
1,21 [0,13 ; 2,28] |
13 % |
0,029 |
|
FAS b (médiane) |
241 |
8,38 |
257 |
10,05 |
1,67 |
20 % |
- |
|
Questionnaire de qualité de vie relatif à la rhinoconjonctivite (RQLQ) |
|||||||
|
FAS b (moyenne ajustée) |
229 |
1,38 |
240 |
1,58 |
0,19 e [0,02 ; 0,37] |
12 % |
0,031 |
|
FAS b (médiane) |
229 |
1,25 |
240 |
1,46 |
0,21 |
14 % |
- |
|
Critères post hoc |
N |
Proportion |
N |
Proportion |
Odds Ratio f (IC à 95 %) |
Valeur de p |
|
|
Pourcentage de jours avec exacerbation de rhinite |
|||||||
|
FAS (estimation) b |
284 |
5,33 % |
298 |
11,14 % |
0,45 [0,28 ; 0,72] |
0,001 |
|
|
Pourcentage de jours avec exacerbation de rhinite malgré la prise de médicaments symptomatiques de la rhinite |
|||||||
|
FAS (estimation) b |
284 |
3,43 % |
298 |
6,50 % |
0,51 [0,32 ; 0,81] |
0,005 |
|
N : Nombre de patients dans le groupe de traitement pour qui des données étaient disponibles pour lanalyse
IC : Intervalle de confiance
a FAS-MI : analyse sur l'ensemble des données disponibles (full analysis set) avec imputations multiples. Pour lanalyse, les patients sortis de létude avant la période dévaluation de lefficacité ont été considérés comme des patients sous placebo. Pour lanalyse principale (FAS-MI), seule la différence absolue a été préspécifiée.
b FAS : analyse sur l'ensemble des données disponibles (full analysis set). Toutes les données disponibles utilisées dans leur intégralité (cest-à-dire conduites chez tous les patients disposant de données au cours de la période dévaluation de lefficacité).
c Différence absolue : placebo moins 12 SQ-HDM, intervalle de confiance à 95 %.
d Différence relative par rapport au placebo : placebo moins 12 SQ-HDM divisé par le placebo.
e La différence entre les groupes 12 SQ-HDM et placebo a été principalement régie par des différences dans les trois domaines suivants : troubles du sommeil, problèmes pratiques et symptômes nasaux.
f Odds ratio pour la survenue dune exacerbation de rhinite : 12 SQ-HDM par rapport au placebo.
Données complémentaires rhinite allergique
Une étude de phase II randomisée, en double aveugle et contrôlée contre placebo a été menée en chambre dexposition aux allergènes chez 124 adultes présentant une rhinite allergique aux acariens. Les sujets ont été sevrés de tout traitement symptomatique anti-allergique avant chaque exposition à lallergène. Lors de lexposition allergénique de fin détude après 24 semaines de traitement par 12 SQ-HDM, 6 SQ-HDM ou placebo, le score moyen des symptômes de rhinite a été de 7,45, IC95% : 6,57 ; 8,33 dans le groupe placebo et de 3,83, IC95% : 2,94 ; 4,72 dans le groupe 12 SQ-HDM, correspondant à une différence absolue de 3,62 et à une différence relative de 49 %, IC95 % : 35 %; 60 %; p < 0,001. La différence entre les groupes 12 SQ-HDM et placebo a été également statistiquement significative à 16 semaines (scores moyens de 5,95 et 8,58, différence absolue de 2,62, différence relative de 30 %, IC95% :17 % ; 42 %, p < 0,001) et à 8 semaines (scores moyens de 6,51 et 8,48, différence absolue de 1,97, différence relative de 20 %, IC95% : 7 % ; 33 %, p = 0,007).
Asthme allergique
Létude MITRA (MT-04)
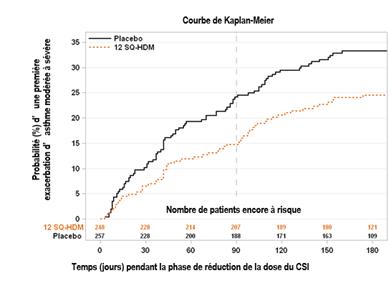
Létude MITRA a été menée chez 834 adultes présentant un asthme allergique aux acariens et insuffisamment contrôlés par la prise quotidienne dun corticostéroïde inhalé (CSI) correspondant à 400 à 1200 µg de budésonide. Tous les patients ont reçu un traitement de 7 à 12 mois par ACARIZAX en complément du corticostéroïde inhalé et d'un bêta2-agoniste inhalé de courte durée daction avant une réduction de la dose du corticostéroïde inhalé. Le protocole n'a pas inclus de phase initiale de recherche de la dose minimale efficace de la corticothérapie inhalée. Lefficacité a été évaluée par le délai de survenue de la première exacerbation dasthme modérée à sévère lors de la période de réduction du corticostéroïde inhalé sur les 6 derniers mois des 13 à 18 mois de traitement.
Une exacerbation dasthme modérée était définie par au moins un des critères suivants et la nécessité de modifier son traitement :
o Réveil(s) nocturne(s) ou augmentation des symptômes : un ou plusieurs réveils nocturnes dus à lasthme et nécessitant la prise dun bêta2-agoniste de courte durée d'action (SABA) lors de deux nuits consécutives, ou augmentation du score quotidien des symptômes ≥ 0,75 pendant deux jours consécutifs par rapport à la valeur initiale à lentrée dans létude.
o Augmentation du nombre de prises dun bêta2-agoniste de courte durée d'action : augmentation du nombre de prises dun bêta2-agoniste de courte durée d'action pendant deux jours consécutifs par rapport à la valeur initiale à lentrée dans létude (augmentation minimale : 4 bouffées/jour).
o Dégradation de la fonction respiratoire : diminution du DEP ≥ 20 % par rapport à la valeur initiale à lentrée dans létude lors de deux matins ou soirs consécutifs, ou diminution du VEMS ≥ 20 % par rapport à la valeur initiale à lentrée dans létude.
o Consultation médicale : consultation dun service durgences ou dun centre investigateur pour traitement de lasthme ne requérant pas une corticothérapie systémique.
Une exacerbation dasthme sévère était définie par la présence dau moins un des deux critères suivants :
o Nécessité dune corticothérapie systémique pendant ≥ 3 jours
o Consultation dun service durgences entraînant une corticothérapie systémique ou une hospitalisation pendant ≥ 12 h.
|
|
Etude MITRA représentation graphique du critère primaire d'efficacité : évolution en fonction du temps du risque dexacerbation dasthme modérée à sévère au cours de la période de réduction ou de sevrage du CSI. Dans le graphique, temps = 0 représente le moment de la réduction de 50 % de la dose du CSI. Après environ 3 mois (soit 90 jours), le traitement par CSI était complétement arrêté chez les patients qui n'avait pas fait dexacerbation.
|
|
Résultats de létude MITRA |
12 SQ-HDM |
Placebo |
Efficacité 12 SQ-HDM par rapport au placebo |
Valeur de p |
|||
|
N |
n (%) |
N |
n (%) |
Risque relatif [IC à 95 %] |
Réduction du risque a |
||
|
Critère primaire |
|||||||
|
Toute exacerbation modérée ou sévère (FAS-MI) b |
282 |
59 (21 %) |
277 |
83 (30 %) |
0,69 [0,50 ; 0,96] |
31 % |
0,027 |
|
Toute exacerbation modérée ou sévère (FAS) c |
248 |
59 (24 %) |
257 |
83 (32 %) |
0,66 [0,47 ; 0,93] |
34 % |
0,017 |
|
Analyses prédéfinies des composantes du critère primaire |
|||||||
|
Réveil(s) nocturne(s) ou augmentation des symptômes c |
248 |
39 (16 %) |
257 |
57 (22 %) |
0,64 [0,42 ; 0,96] |
36 % |
0,031 |
|
Augmentation de lutilisation dun SABA c |
248 |
18 (7 %) |
257 |
32 (12 %) |
0,52 [0,29 ; 0,94] |
48 % |
0,029 |
|
Dégradation de la fonction respiratoire c |
248 |
30 (12 %) |
257 |
45 (18 %) |
0,58 [0,36 ; 0,93] |
42 % |
0,022 |
|
Exacerbation sévère c |
248 |
10 (4 %) |
257 |
18 (7 %) |
0,49 [0,23 ; 1,08] |
51 % |
0,076 |
|
N : Nombre de patients dans le groupe de traitement pour qui des données étaient disponibles pour lanalyse. n (%) : nombre et pourcentage de patients du groupe de traitement répondant au critère. IC : Intervalle de confiance a Estimation par le risque relatif. b FAS-MI : analyse sur l'ensemble des données disponibles (full analysis set) avec imputations multiples. Pour lanalyse, les patients sortis de létude avant la période dévaluation de lefficacité ont été considérés comme des patients sous placebo. c FAS : analyse sur l'ensemble des données disponibles (full analysis set). Toutes les données disponibles utilisées dans leur intégralité (tous les patients chez qui des données avaient été recueillies au cours de la période dévaluation de lefficacité).
|
|||||||
Des analyses des symptômes dasthme et de la prise de médicaments symptomatiques au cours des 4 dernières semaines de la période de traitement avant la réduction de la dose du corticostéroïde inhalé ont été également menées a posteriori afin dévaluer leffet dACARIZAX en complément dun corticostéroïde inhalé. Ces analyses ont porté sur les scores des symptômes d'asthme diurnes et nocturnes, les réveils nocturnes et les prises dun bêta2-agoniste de courte durée d'action. Elles ont montré des différences numériques constamment en faveur du groupe 12 SQ-HDM comparativement au groupe placebo pour tous les paramètres évalués durant les 4 semaines précédant la réduction de la dose du corticostéroïde inhalé. Les différences ont été statistiquement significatives uniquement pour le score des symptômes d'asthme diurnes (p = 0,0450) et lodds radio pour labsence de réveil nocturne (p = 0,0409).
Données complémentaires asthme allergique
Lors dune étude de phase II randomisée, en double aveugle et contrôlée contre placebo, 604 patients ≥ 14 ans présentant un asthme allergique aux acariens contrôlé par la prise dun corticostéroïde inhalé (100 à 800 µg de budésonide) et ayant des antécédents de rhinite allergique aux acariens ont été randomisés pour recevoir 1, 3 ou 6 SQ-HDM ou un placebo pendant environ un an. Lors de la période dévaluation de lefficacité au cours des 4 dernières semaines de l'étude, la modification moyenne de la dose du corticostéroïde inhalé par rapport à la valeur initiale à lentrée dans l'étude a été de 207,6 µg de budésonide dans le groupe 6 SQ-HDM et 126,3 µg dans le groupe placebo, correspondant à une différence absolue de 81 µg de budésonide par jour ([27 ; 136] intervalle de confiance à 95 %, p = 0,004). Les réductions relatives moyennes et médianes de la dose du corticostéroïde inhalé par rapport à la valeur initiale à lentrée dans létude ont été de 42 % et 50 % dans le groupe 6 SQ-HDM et de 15 % et 25 % dans le groupe placebo. Une analyse dun sous-groupe (N = 108) de patients chez qui le contrôle de lasthme était moindre et qui prenaient un corticostéroïde inhalé à raison de ≥ 400 µg de budésonide a été conduite a posteriori et a montré que la modification moyenne de la dose quotidienne du corticosteroide inhalé par rapport à la valeur initiale à lentrée dans létude était de 384,4 µg de budésonide dans le groupe 6 SQ-HDM et de 57,8 µg dans le groupe placebo, correspondant à une différence absolue de 327 µg de budésonide par jour IC95% : 182 ; 471, p < 0,0001, analyse a postériori).
Population pédiatrique
ACARIZAX nest pas indiqué chez les patients de moins de 18 ans (voir rubrique 4.2).
Les données sur la sécurité demploi et la tolérance chez des patients âgés de 5 à 17 ans sont limitées.
LAgence européenne des médicaments a accordé une dérogation à lobligation de soumettre les résultats détudes réalisées avec ACARIZAX chez des enfants âgés de moins de 5 ans dans lallergie respiratoire aux acariens (traitement de la rhinite allergique, prévention de lasthme, traitement de lasthme).
LAgence européenne des médicaments a différé lobligation de soumettre les résultats détudes additionnelles réalisées avec ACARIZAX chez des enfants âgés de 5 ans ou plus dans lallergie respiratoire aux acariens (traitement de la rhinite allergique, prévention de lasthme, traitement de lasthme).
Patients âgés
ACARIZAX nest pas indiqué chez les patients de plus de 65 ans (voir rubrique 4.2). Il existe des données limitées sur la sécurité demploi et la tolérance chez des patients de plus de 65 ans.
Traitement à long terme
Les recommandations thérapeutiques internationales préconisent une durée d'immunothérapie allergénique d'environ 3 ans pour modifier lévolution de la maladie. Des données defficacité issues de létude MITRA sont disponibles pour un traitement de 18 mois par ACARIZAX. Lefficacité à long terme na pas été établie.
5.2. Propriétés pharmacocinétiques
Les molécules actives dun extrait allergénique sont essentiellement composées de protéines. Dans le cas de limmunothérapie allergénique administrée par voie sublinguale, des études ont montré labsence dabsorption passive des allergènes à travers la muqueuse buccale. Des données indiquent que les allergènes seraient captés à travers la muqueuse buccale par les cellules dendritiques, en particulier les cellules de Langerhans. Les allergènes non absorbés de cette façon seraient hydrolysés en acides aminés et en petits polypeptides dans la lumière des voies digestives. Aucune donnée ne suggère que les allergènes présents dans ACARIZAX soient significativement absorbés dans le système vasculaire après administration sublinguale.
5.3. Données de sécurité préclinique
Mannitol
Hydroxyde de sodium (pour ajustement du pH)
6.4. Précautions particulières de conservation
Pas de précaution particulière de conservation.
6.5. Nature et contenu de l'emballage extérieur
Boîtes de 10, 30 et 90.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
BØGE ALLE 6-8
2970 HØRSHOLM
DANEMARK
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 300 453 3 6 : boîte de 10 lyophilisats oraux de 12 SQ-HDM sous plaquette thermoformée (Alu/Alu).
· 34009 300 453 4 3 : boîte de 30 lyophilisats oraux de 12 SQ-HDM sous plaquette thermoformée (Alu/Alu).
· 34009 550 167 3 1 : boîte de 90 lyophilisats oraux de 12 SQ-HDM sous plaquette thermoformée (Alu/Alu).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[A compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[A compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.