RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 05/07/2016
NOCDURNA 50 microgrammes, lyophilisat oral
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour la liste complète des excipients, voir rubrique 6.1.
Lyophilisat oral, blanc, rond, denviron 12 mm, comportant l'inscription 50 sur une face.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
· Femmes : 25 microgrammes par jour, une heure avant le coucher, administrés par voie sublinguale sans eau.
· Hommes : 50 microgrammes par jour, une heure avant le coucher, administrés par voie sublinguale sans eau.
Il nest pas recommandé daugmenter la dose de ce produit chez les patients âgés de 65 ans et plus.
Si des doses supérieures sont envisagées pour les patients âgés de moins de 65 ans en cas de réponse insuffisante à Nocdurna, il convient d'utiliser d'autres dosages de lyophilisats oraux de desmopressine (voir rubriques 4.4, 4.8 et 5.1).
En cas de signes ou symptômes évocateurs d'une rétention hydrique et/ou d'une hyponatrémie (céphalées, nausées/vomissements, prise de poids, et convulsions dans les cas sévères), le traitement doit être interrompu et réévalué. Si le traitement est repris, la restriction hydrique devra être plus stricte et la natrémie sera surveillée (voir rubrique 4.4).
Le traitement par Nocdurna doit être arrêté si la natrémie chute au-dessous de la limite inférieure de la normale (cest-à-dire 135 mmol/L).
Populations particulières
Patients âgés (65 ans et plus)
Les patients âgés présentent un risque accru dhyponatrémie avec le traitement par desmopressine et sont également susceptibles de présenter une insuffisance rénale. Par conséquent, il convient de faire preuve de prudence dans ce groupe dâge et les doses quotidiennes supérieures à 25 microgrammes chez les femmes et 50 microgrammes chez les hommes doivent être évitées. Chez les patients âgés de 65 ans et plus, les taux sériques de sodium doivent se situer dans les limites de la normale, avant linitiation du traitement, durant la première semaine (après 4 à 8 jours de traitement) et après 1 mois de traitement. Il convient d'arrêter Nocdurna si la natrémie chute au-dessous de la limite inférieure de la normale (voir rubrique 4.4).
La poursuite du traitement doit être envisagée avec prudence chez les patients âgés ne montrant pas de bénéfice thérapeutique au-delà de 3 mois.
Insuffisance rénale
Nocdurna est contre-indiqué chez les patients présentant une insuffisance rénale modérée ou sévère (voir rubrique 4.3).
Insuffisance hépatique
Aucune adaptation posologique nest nécessaire chez les patients présentant une insuffisance hépatique (voir rubrique 5.2).
Population pédiatrique
Nocdurna nest pas indiqué chez lenfant pour le traitement symptomatique de la nycturie associée à une polyurie nocturne idiopathique.
Mode dadministration
Nocdurna est placé sous la langue, où il se dissout sans eau.
Lapport alimentaire peut réduire lintensité et la durée de leffet antidiurétique de la desmopressine administrée à faibles doses (voir rubrique 5.2).
· Hypersensibilité à la substance active ou à lun des excipients mentionnés à la rubrique 6.1.
· Polydipsie ou potomanie (conduisant à une production durine supérieure à 40 ml/kg/24 heures).
· Insuffisance cardiaque connue ou suspectée, ou autre affection associée à une surcharge hydrique suffisante pour nécessiter un traitement par diurétiques, y compris des antécédents de telles affections.
· Insuffisance rénale modérée et sévère (clairance de la créatinine inférieure à 50 ml/min).
· Antécédents connus dhyponatrémie.
· Syndrome de sécrétion inappropriée d'hormone antidiurétique.
4.4. Mises en garde spéciales et précautions d'emploi
Il est impératif de restreindre toute prise de liquides au moins 1 heure avant et pendant les 8 heures suivant ladministration. Un traitement par desmopressine sans diminution parallèle de la prise de liquide peut entraîner une rétention hydrique prolongée et/ou une hyponatrémie, avec ou sans survenue de symptômes d'alarme (tels que céphalées, nausées/vomissements, prise de poids, voire convulsions dans les cas sévères).
Les patients âgés ayant une natrémie située dans la limite inférieure de la normale présentent potentiellement un risque accru dhyponatrémie. Chez les patients âgés de 65 ans et plus, la natrémie doit être surveillée avant linitiation du traitement, durant la première semaine du traitement (4 à 8 jours) et de nouveau un mois après le début du traitement (voir rubrique 4.2).
A la dose de 50 microgrammes, il se peut que les femmes présentent un risque supérieur dhyponatrémie comparativement aux hommes (voir rubrique 5.1). Il est donc important de respecter les recommandations posologiques spécifiques au sexe.
Il convient d'arrêter Nocdurna si la natrémie chute au-dessous de la limite inférieure de la normale.
La desmopressine doit être utilisée avec prudence chez les patients présentant une affection caractérisée par un déséquilibre hydrique et/ou électrolytique.
Le traitement par desmopressine doit être interrompu et réévalué en cas de maladie intercurrente aiguë caractérisée par un déséquilibre hydrique et/ou électrolytique (comme des infections systémiques, de la fièvre, une gastro-entérite).
Des précautions visant à éviter une hyponatrémie incluant une restriction hydrique stricte et une surveillance plus fréquente de la natrémie, devront être respectées, en cas de traitement concomitant par des médicaments connus pour induire un syndrome de sécrétion inappropriée d'hormone anti-diurétique, par exemple les antidépresseurs tricycliques, les antidépresseurs inhibiteurs sélectifs de la recapture de la sérotonine, la chlorpromazine, les diurétiques et la carbamazépine, et certains antidiabétiques appartenant au groupe des sulfamides hypoglycémiants, en particulier le chlorpropamide, et en cas de traitement concomitant par des médicaments anti-inflammatoires non stéroïdiens (AINS).
Une prudence particulière est requise chez les patients prenant des diurétiques thiazidiques ou des diurétiques de lanse pour une hypertension artérielle ou d'autres affections non associées à une surcharge hydrique. La surveillance de la natrémie est recommandée chez ces patients.
Une dysfonction vésicale sévère et une obstruction vésicale doivent être écartées avant de débuter le traitement.
La desmopressine doit être administrée avec prudence en cas de mucoviscidose, d'insuffisance coronarienne, d'hypertension artérielle, d'insuffisance rénale chronique et de pré-éclampsie.
Le diagnostic de diabète insipide néphrogènique doit être envisagé en l'absence de réduction des mictions nocturnes après le commencement dun traitement par la desmopressine.
Une prudence particulière est requise chez les patients prenant du lithium, car il existe un risque que le diabète insipide néphrogènique de stade précoce induit par le lithium soit masqué par ladministration de desmopressine pour lindication de nycturie. La desmopressine nest pas recommandée en cas de suspicion de diabète insipide néphrogènique induit par le lithium.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Interactions pharmacodynamiques
Les substances connues pour induire un syndrome de sécrétion inappropriée d'hormone antidiurétique peuvent être associées à un risque accru de rétention hydrique/hyponatrémie (par exemple, antidépresseurs tricycliques, antidépresseurs inhibiteurs sélectifs de la recapture de la sérotonine, chlorpromazine, diurétiques et carbamazépine, ainsi que certains antidiabétiques appartenant au groupe des sulfamides hypoglycémiants, en particulier le chlorpropamide) (voir rubrique 4.4).
Les AINS et loxytocine peuvent potentialiser leffet antidiurétique de la desmopressine et induire une rétention hydrique/hyponatrémie (voir rubrique 4.4).
Le lithium peut diminuer leffet antidiurétique.
Interactions pharmacocinétiques
Le traitement concomitant par lopéramide peut entraîner une multiplication par 3 des concentrations plasmatiques de desmopressine après administration orale, ce qui peut augmenter le risque de rétention hydrique/dhyponatrémie. Bien que cela nait pas été étudié, d'autres médicaments ralentissant le transit intestinal peuvent avoir le même effet.
Un repas standardisé à 27 % de matières grasses a réduit significativement labsorption (vitesse et ampleur) des comprimés de desmopressine. Il n'a pas été observé deffet significatif sur la pharmacodynamie (production durine ou osmolalité).
Lapport alimentaire peut réduire lintensité et la durée de leffet antidiurétique de la desmopressine en comprimé, administrée à faibles doses.
4.6. Fertilité, grossesse et allaitement
Grossesse
La desmopressine doit être prescrite avec prudence aux femmes enceintes.
Les données concernant un nombre limité (n = 53) de grossesses exposées chez des femmes atteintes de diabète insipide, ainsi que des données sur un nombre limité de grossesses exposées chez des femmes ayant des complications hémorragiques (n = 216), indiquent l'absence deffets indésirables de la desmopressine sur la grossesse ou sur la santé du ftus/nouveau-né. À ce jour, aucune donnée épidémiologique pertinente nest disponible. Les études menées chez lanimal nindiquent pas deffets nocifs directs ou indirects en ce qui concerne la gestation, le développement embryonnaire/ftal, la parturition et le développement postnatal.
Les études de reproduction chez lanimal nont pas montré deffets cliniquement pertinents sur les parents ni sur la descendance. Lanalyse in vitro de modèles de cotylédons humains a montré labsence de transport transplacentaire de la desmopressine lors de l'administration à une concentration thérapeutique correspondant à la dose recommandée.
Les résultats de lanalyse du lait de mères allaitantes recevant de l'acétate de desmopressine à dose élevée (300 microgrammes par voie intranasale) indiquent que les quantités de desmopressine susceptibles dêtre transférées à lenfant sont nettement inférieures aux quantités pouvant influencer la diurèse. Par conséquent, il nest pas considéré comme nécessaire d'arrêter lallaitement.
Fertilité
Les études avec la desmopressine chez les animaux nont montré aucune altération de la fertilité chez les rats mâles et femelles.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les événements indésirables les plus fréquemment rapportés dans les études cliniques de Nocdurna dans lindication nycturie conduites chez des sujets de sexe masculin (50 microgrammes ; N =222) et féminin (25 microgrammes ; N = 219), étaient : sécheresse de la bouche (13 %), céphalée (3 %), hyponatrémie (3 %) et sensation vertigineuse (2 %).
Description deffets indésirables dintérêt particulier :
Leffet indésirable le plus grave avec la desmopressine est lhyponatrémie, qui est associée à des céphalées, des nausées, des vomissements, une diminution du taux de sodium sérique, une prise de poids, une perte de connaissance, des douleurs abdominales, des crampes musculaires, des sensations vertigineuses, une confusion, une diminution de l'état de conscience et, dans les cas sévères, des convulsions et un coma. Lhyponatrémie est une conséquence de l effet antidiurétique provenant dune réabsorption accrue de leau par les tubules rénaux et dune dilution osmotique du plasma. Dans les études menées chez des sujets adultes traités pour nycturie, la majorité des sujets a développé une hyponatrémie au cours des premiers jours du traitement ou après une augmentation de la dose. Une attention particulière doit être portée aux précautions indiquées dans la rubrique 4.4.
Les femmes présentent un risque supérieur dhyponatrémie, qui pourrait être dû à une sensibilité plus élevée des tubules rénaux à la vasopressine et à ses analogues chez les femmes comparativement aux hommes. Les recommandations posologiques plus faibles chez les femmes visent à réduire ce risque. Le risque d'hyponatrémie chez les sujets âgés de 65 ans et plus, est réduit par la surveillance de la natrémie dans ce groupe dâge (voir rubriques 4.2 et 4.4).
Liste des effets indésirables
Le tableau 1 ci-dessous indique les fréquences des réactions indésirables rapportées. Les fréquences sont définies comme suit : très fréquent (≥1/10), fréquent (≥1/100 à <1/10) et peu fréquent (≥1/1 000 à <1/100).
Tableau 1 : Fréquence des effets indésirables rapportés (lors des études de Phase III et après commercialisation)
|
Base de données MedDRA des classes de systèmes dorganes |
Très fréquent (≥ 1/10) |
Fréquent (≥ 1/100 à < 1/10) |
Peu fréquent (≥ 1/1 000 à < 1/100)
|
|
Troubles du métabolisme et de la nutrition |
|
Hyponatrémie |
|
|
Affections du système nerveux |
|
Céphalées Sensation vertigineuse |
|
|
Affections gastro-intestinales
|
Sécheresse de la bouche* |
Nausées Diarrhée |
Constipation Douleurs abdominales |
|
Troubles généraux et anomalies au site dadministration |
|
|
Fatigue dèmes périphériques |
*Il est à noter que dans certaines des études cliniques, les sujets étaient spécifiquement interrogés sur la sécheresse buccale.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Un surdosage de Nocdurna conduit à une durée d'action prolongée, augmentant ainsi le risque de rétention hydrique et dhyponatrémie.
Traitement :
Le traitement de l'hyponatrémie doit être individualisé. Les recommandations générales suivantes peuvent toutefois être données. Lhyponatrémie est traitée par interruption du traitement par desmopressine, restriction hydrique et traitement symptomatique si nécessaire.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Vasopressine et analogues. Code ATC : H01B A02
Mécanisme daction
Nocdurna contient de la desmopressine, qui est un analogue de synthèse de l'hormone antidiurétique naturelle, l'arginine-vasopressine (AVP). La desmopressine reproduit leffet antidiurétique de la vasopressine, se liant aux récepteurs V2 dans les tubules collecteurs rénaux pour provoquer une réabsorption de leau dans le corps. Cette réabsorption diminue à son tour la production durine pendant la nuit. En raison des doses faibles proposées, selon le sexe (25 microgrammes pour les femmes et 50 microgrammes pour les hommes), et de la durée d'action limitée de Nocdurna, lactivité antidiurétique est limitée à la période de sommeil nocturne.
Effets pharmacodynamiques
Dans l'étude CS29, la dose de Nocdurna corrigée en fonction du poids ayant induit un effet du médicament équivalent à 50 % de leffet maximal pouvant être atteint sur le volume nocturne durine différait significativement entre les femmes et les hommes. La valeur dexposition estimée était 2,7 fois supérieure (IC à 95 % : 1,3-8,1) chez les hommes par rapport aux femmes pour obtenir un effet dynamique identique, correspondant à une sensibilité supérieure à la desmopressine chez les femmes. Le développement dune hyponatrémie est dépendant de la dose. Le risque de développement dune hyponatrémie est plus élevé chez les femmes que chez les hommes. Lincidence de lhyponatrémie augmente avec lâge (voir rubriques 4.2 et 4.4).
Efficacité et sécurité clinique
Lefficacité de Nocdurna a été démontrée dans deux études randomisées en double aveugle contrôlées versus placebo chez respectivement 268 femmes (étude CS40, desmopressine lyophilisat oral 25 microgrammes versus placebo) et 395 hommes (étude CS41, desmopressine lyophilisat oral 50 microgrammes et 75 microgrammes versus placebo) présentant une nycturie définie comme une moyenne de 2 mictions nocturnes et plus par nuit, et une polyurie chez 90 % des femmes et 87 % des hommes.
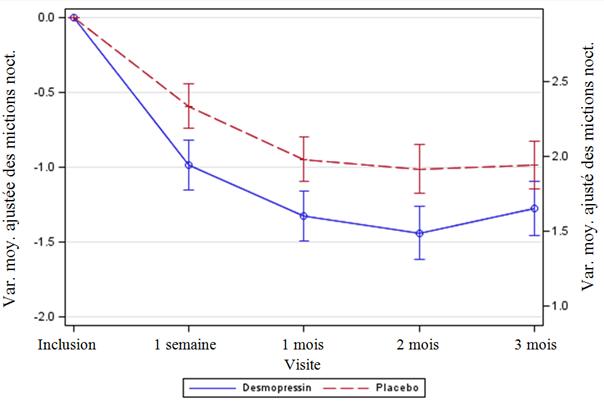
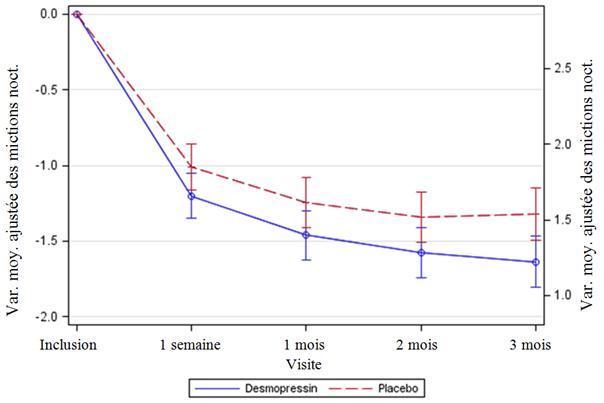
Les deux études ont atteint les 2 co-critères primaires d'évaluation, avec des différences statistiquement significatives en faveur de la desmopressine lyophilisat oral sur la période de 3 mois. Il a été observé une diminution statistiquement significative du nombre moyen ajusté de mictions nocturnes à partir de linclusion sous desmopressine lyophilisat oral 25 microgrammes (-1,46) comparativement au placebo (-1,24) dans l'étude chez les femmes (p =0,028) (Fig. 1) et sous desmopressine lyophilisat oral 50 microgrammes (-1,25) comparativement au placebo (-0,88) dans l'étude chez les hommes (p = 0,0003) (Fig. 2). La proportion de sujets présentant une diminution >33 % du nombre moyen de mictions nocturnes (répondeurs) a été significativement augmentée, presque doublée. Lodds ratio de la diminution >33 % avec la desmopressine lyophilisat oral 25 microgrammes comparativement au placebo était de 1,85 (p = 0,006) dans l'étude menée chez les femmes et lodds ratio de la diminution >33 % avec la desmopressine lyophilisat oral 50 microgrammes comparativement au placebo était de 1,98 (p = 0,0009) dans l'étude menée chez les hommes.
Pour les critères d'évaluation secondaires, entre linclusion et 3 mois de traitement, il a été observé une augmentation de la première période de sommeil non interrompu/du temps avant la première miction, avec une différence deffet de 49 minutes chez les femmes et de 39 minutes chez les hommes. Il a été observé une amélioration statistiquement significative de la qualité de vie avec la desmopressine lyophilisat oral 25 microgrammes (score total N-QoL 27,24) comparativement au placebo (21,90) (p = 0,0226) chez les femmes et une amélioration avec la desmopressine lyophilisat oral 50 microgrammes (score total N-QoL 18,37) comparativement au placebo (13,88) (p = 0,0385) chez les hommes. Dans les deux études, une association significative (p < 0,0001) a été observée entre la réponse au traitement (réduction du nombre de mictions nocturnes et augmentation de la première période de sommeil non interrompu) et les améliorations de la qualité de vie des patients.
Figure 1. Co-critère primaire d'évaluation : Variation moyenne ajustée par rapport à linclusion des mictions nocturnes durant les 3 mois de traitement (Femmes, ensemble d'analyse intégral CS40)
|
|
|
Figure 2. Co-Critère primaire d'évaluation : Variation moyenne ajustée par rapport à linclusion des mictions nocturnes durant les 3 mois de traitement (Hommes, ensemble d'analyse intégral CS41)
|
|
Dans une étude clinique randomisée en double aveugle, lefficacité et la tolérance dun traitement associant la desmopressine lyophilisat oral et la toltérodine en gélules à libération prolongée ont été examinées pour le traitement de lhyperactivité vésicale avec nycturie chez les femmes, pendant une période de 3 mois. Quarante-neuf sujets ont été exposés à une association de Nocdurna (desmopressine lyophilisat oral) 25 microgrammes et de toltérodine 4 milligrammes. Aucun effet indésirable grave n'a été observé dans cette étude et le profil de tolérance du traitement combiné était similaire au profil de tolérance de Nocdurna 25 microgrammes. Lefficacité en termes de réduction du nombre moyen de mictions nocturnes par rapport à linclusion durant le traitement de 3 mois était numériquement supérieure dans le groupe de traitement d'association versus le groupe de toltérodine en monothérapie (contraste entre traitements, -0,34 miction) dans lensemble d'analyse intégral, et la différence a atteint la signification statistique (p = 0,049) avec un contraste entre traitements de -0,41 miction dans lensemble d'analyse per protocole.
Différences de tolérance clinique et defficacité entre sexes
Létude clinique [FE992026 CS029] a analysé la relation dose-réponse à Nocdurna chez des femmes et des hommes à des doses comprises entre 10 et 100 microgrammes : chez les femmes, il n'a pas été observé de gain supplémentaire de leffet pharmacodynamique au-delà de la dose de 25 microgrammes, ce qui indique que le plateau de la relation dose-réponse était atteint à 25 microgrammes chez les femmes. Chez les hommes, la réduction du volume urinaire était supérieure à la dose de 50 microgrammes, mais pas sensiblement supérieure à la dose de 100 microgrammes. Laugmentation des doses à 50 microgrammes chez les femmes n'a pas conduit à une efficacité supérieure, mais a été associée à une multiplication par 5 du risque dhyponatrémie comparativement aux hommes dans le groupe d'âge des plus de 50 ans (p = 0,015).
5.2. Propriétés pharmacocinétiques
La biodisponibilité absolue moyenne globale de la desmopressine administrée par voie sublinguale à partir de précédentes études de recherche de doses de 200, 400 et 800 microgrammes est de 0,25 %, avec un intervalle de confiance à 95 % compris entre 0,21 et 0,31 %. La desmopressine présente une variabilité intra et inter individuelle modérée à élevée en termes de biodisponibilité. La desmopressine montre une linéarité à la dose en ce qui concerne lASC et la Cmax dans lintervalle compris entre 60 et 240 microgrammes. Toutefois, la biodisponibilité des doses inférieures à 60 microgrammes n'a pas été évaluée.
Distribution
La distribution de la desmopressine est décrite au mieux par un modèle de distribution à deux compartiments, avec un volume de distribution durant la phase d'élimination compris entre 0,3 et 0,5 L/kg.
Biotransformation
Le métabolisme in vivo de la desmopressine na pas été étudié. Les études de métabolisme de la desmopressine in vitro dans des microsomes hépatiques ont montré que la quantité métabolisée dans le foie par le système du cytochrome P450 nest pas significative. Par conséquent, le métabolisme hépatique humain in vivo par le système du cytochrome P450 est improbable. Il est probable que leffet de la desmopressine sur la pharmacocinétique des autres médicaments soit minimal, en raison de labsence dinhibition du système de métabolisation des médicaments du cytochrome P450.
Élimination
La clairance totale de la desmopressine a été calculée à 7,6 l/h. La demi-vie terminale de la desmopressine est estimée à 2,8 heures. Chez les sujets sains, la fraction excrétée sous forme inchangée était de 52 % (44 % - 60 %).
Linéarité/non-linéarité
Il nexiste pas d'indications de non-linéarité, pour aucun des paramètres pharmacocinétiques de la desmopressine.
Caractéristiques dans des groupes spécifiques de patients
Insuffisance rénale :
LASC et la demi-vie augmentent avec la sévérité de l'insuffisance rénale. La desmopressine est contre-indiquée chez les patients présentant une insuffisance rénale modérée et sévère (clairance de la créatinine inférieure à 50 ml/min).
Tableau 2 : Paramètres pharmacocinétiques pour les différents degrés d'insuffisance rénale. Données de létude CS001.
|
|
Clairance de la créatinine |
Fonction rénale |
ASC (h*pg/mL) |
T½ (h) |
|
Sain |
>80 mL/min |
Normale |
186 |
2,8 |
|
Léger |
50-80 mL/min |
Insuffisance légère |
281 |
4,0 |
|
Modéré |
30-49 mL/min |
Insuffisance modérée |
453 |
6,7 |
|
Sévère |
5-29 mL/min |
Insuffisance sévère |
682 |
8,7 |
Insuffisance hépatique :
Il na pas été conduit d'études dans cette population.
Il est improbable que la desmopressine interagisse avec les médicaments influant sur le métabolisme hépatique, car il a été montré que la desmopressine ne subit pas de métabolisme hépatique significatif dans des études in vitro avec des microsomes humains.
5.3. Données de sécurité préclinique
Il n'a pas été conduit d'études de carcinogénicité avec la desmopressine, car elle très proche de l'hormone peptidique naturelle.
Mannitol (E 421)
Acide citrique, anhydre
6.4. Précautions particulières de conservation
Pas dexigence particulière en ce qui concerne la température de conservation.
À conserver dans son emballage d'origine, à l'abri de l'humidité et de la lumière.
Utiliser immédiatement après ouverture dune alvéole individuelle.
6.5. Nature et contenu de l'emballage extérieur
Plaquettes thermoformées prédécoupées contenues dans un étui.
Le film en aluminium alvéolaire (10 alvéoles) et la feuille de recouvrement sont des films multicouches stratifiés thermoscellés respectivement en PVC/OPA/alu/OPA/PVC et Alu/PET/papier. Boites de 10, 30, 90 ou 100 lyophilisats oraux
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Pas dexigences particulières.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
7, rue Jean-Baptiste Clément
94250 GENTILLY
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 300 610 3 9 : 30 lyophilisats oraux sous plaquette (PVC/OPA/alu/OPA/PVC et Alu/PET/papier ) . Boite de 3 plaquettes.
· 34009 550 221 1 4 : 90 lyophilisats oraux sous plaquette (PVC/OPA/alu/OPA/PVC et Alu/PET/papier ) . Boite de 9 plaquettes.
· 34009 550 221 2 1 : 100 lyophilisats oraux sous plaquette (PVC/OPA/alu/OPA/PVC et Alu/PET/papier ) . Boite de 10 plaquettes
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II