RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 10/08/2016
ILUVIEN 190 microgrammes, implant intravitréen avec applicateur
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque implant intravitréen contient 190 microgrammes dacétonide de fluocinolone.
Pour la liste complète des excipients, voir rubrique 6.1.
Implant intravitréen avec applicateur.
Cylindre de couleur brun clair mesurant environ 3,5 mm x 0,37 mm.
Applicateur avec aiguille 25 G.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
La dose recommandée est un implant dILUVIEN dans lil atteint. Ladministration simultanée dans les deux yeux nest pas recommandée (voir rubrique 4.4).
Chaque implant ILUVIEN libère de lacétonide de fluocinolone pendant une durée allant jusquà 36 mois. Un autre implant peut être administré après 12 mois si le patient présente une perte dacuité visuelle ou une augmentation de lépaisseur rétinienne secondaire à une récidive ou à une aggravation de ldème maculaire diabétique (voir rubrique 5.1).
Un retraitement par ILUVIEN ne doit être envisagé que si les bénéfices potentiels sont supérieurs aux risques.
Le traitement par ILUVIEN est réservé aux patients présentant une réponse insuffisante à un traitement antérieur par photocoagulation au laser ou aux autres traitements disponibles pour ldème maculaire diabétique.
Population pédiatrique
Il ny a pas dutilisation justifiée de lacétonide de fluocinolone administré par voie intravitréenne dans la population pédiatrique dans lindication ddème maculaire diabétique (OMD).
Populations particulières
Aucune adaptation posologique nest nécessaire chez les patients âgés ou chez les patients présentant une insuffisance rénale ou hépatique.
Mode d'administration
VOIE INTRAVITREENNE UNIQUEMENT.
ILUVIEN ne doit être administré que par voie intravitréenne et par un ophtalmologiste expérimenté dans les injections intravitréennes.
La procédure dinjection intravitréenne doit être réalisée en conditions dasepsie contrôlées, incluant le port de gants stériles, lutilisation dun champ stérile et dun blépharostat (ou équivalent) stérile. Une anesthésie adéquate et une antisepsie antibactérienne à large spectre doivent être administrées avant linjection.
La procédure dinjection dILUVIEN est la suivante :
1. Un collyre antibiotique peut être administré avant lintervention à lappréciation de lophtalmologiste traitant.
2. Juste avant linjection, instiller une goutte danesthésique local sur le site dinjection (le quadrant temporal inférieur est recommandé), suivie de lapplication dun coton‑tige imbibé danesthésique ou dune injection sous‑conjonctivale dun anesthésique approprié.
3. Instiller 2 ou 3 gouttes dun antiseptique local approprié dans le cul‑de‑sac conjonctival. Les paupières peuvent être nettoyées avec des cotons-tiges imbibés dun antiseptique local approprié. Placer un blépharostat stérile. Demander au patient de regarder vers le haut et appliquer un coton‑tige imbibé dun antiseptique approprié sur le site dinjection. Laisser lantiseptique local sécher pendant 30 à 60 secondes avant linjection dILUVIEN.
4. Lextérieur de la plaquette thermoformée ne doit pas être considéré comme stérile. Une assistant(e) (en conditions non stériles) doit sortir la plaquette thermoformée de la boîte puis examiner la plaquette et le film pour vérifier labsence de dommage. En cas de dommage, ne pas utiliser lunité.
Si lunité peut être utilisée, lassistant(e) doit retirer le film de la plaquette sans toucher la surface intérieure.
5. Vérifier par la fenêtre de visualisation de lapplicateur préchargé que celui‑ci contient bien limplant.
6. En portant des gants stériles, retirer lapplicateur de la plaquette thermoformée en ne touchant que la surface stérile et lapplicateur.
Le capuchon de protection de laiguille ne doit être retiré que lorsque limplant ILUVIEN est prêt à être injecté.
Avant linjection, lembout de lapplicateur doit être maintenu incliné pour garantir que limplant est positionné correctement dans lapplicateur.
7. La procédure dinjection doit être effectuée en deux étapes pour limiter la quantité dair administrée avec limplant. Avant dinsérer laiguille dans lil, appuyer sur le bouton et le faire glisser jusquau premier arrêt (au niveau des repères noirs incurvés le long de la glissière du bouton). Au premier arrêt, relâcher le bouton, il passera en position UP (HAUT). Si le bouton ne passe pas en position UP, ne pas continuer à utiliser cette unité.
8. Le positionnement optimal de limplant est la région située sous la papille optique et à larrière de léquateur de lil. A laide dun compas, mesurer une distance de 4 mm à partir du limbe dans le quadrant temporal inférieur.
9. Retirer avec précaution le capuchon de protection de laiguille et vérifier que la pointe de laiguille nest pas courbée.
10. Déplacer doucement la conjonctive de façon à ce que, après le retrait de laiguille, les sites dinsertion conjonctival et scléral de laiguille ne soient pas alignés. Prendre des précautions pour éviter tout contact entre laiguille et le bord de la paupière ou les cils. Insérer laiguille dans lil. Pour libérer limplant, le bouton étant en position UP, faire glisser le bouton vers lavant jusquà lextrémité de sa glissière et retirer laiguille. Remarque : vérifier que le bouton a atteint lextrémité de la glissière avant de retirer laiguille.
11. Retirer le blépharostat et vérifier par ophtalmoscopie indirecte le positionnement de limplant, la perfusion correcte de lartère rétinienne et labsence de toute autre complication. La visualisation de limplant peut être facilitée en appuyant sur la sclérotique. Lexamen doit inclure un contrôle de la perfusion de la tête du nerf optique immédiatement après linjection. Une mesure immédiate de la PIO peut être effectuée à lappréciation de lophtalmologiste.
Après lintervention, les patients doivent être surveillés pour détecter des complications éventuelles telles quendophtalmie, augmentation de la pression intraoculaire, décollement de la rétine et hémorragies ou décollements vitréens. Une biomicroscopie avec tonométrie doit être réalisée dans les deux à sept jours suivant linjection de limplant.
Compte tenu de la durée de libération de lacétonide de fluocinolone (environ 36 mois), il est recommandé de poursuivre ces contrôles au moins une fois par trimestre afin de détecter lapparition déventuelles complications (voir rubrique 4.4).
Limplant intravitréen ILUVIEN est contre-indiqué en cas de glaucome préexistant ou dinfection oculaire ou périoculaire active ou suspectée, incluant la plupart des maladies virales de la cornée et de la conjonctive, dont la kératite épithéliale active à Herpes simplex (kératite dendritique), la vaccine, la varicelle, les infections mycobactériennes et les mycoses.
ILUVIEN est contre-indiqué chez les patients présentant une hypersensibilité à la substance active ou à lun des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Ladministration de corticoïdes intravitréens peut provoquer une cataracte, une augmentation de la pression intraoculaire, un glaucome et majorer le risque dinfections secondaires.
La sécurité et lefficacité dILUVIEN administré simultanément dans les deux yeux nont pas été étudiées. Il est recommandé de ne pas injecter un implant dans chaque il lors de la même séance de traitement. Le traitement simultané des deux yeux nest pas recommandé tant que la réponse oculaire et systémique au premier implant nest pas connue (voir rubrique 4.2).
Dans les études FAME, une chirurgie de la cataracte a été réalisée chez 80 % des patients phaques traités par lacétonide de fluocinolone (voir rubrique 4.8). Les patients phaques doivent être surveillés étroitement pour détecter des signes de cataracte après le traitement.
Dans les études FAME, 38 % des patients traités par lacétonide de fluocinolone ont eu besoin dun traitement hypotonisant (voir rubrique 4.8). Lacétonide de fluocinolone doit être utilisé avec prudence chez les patients ayant une PIO initiale élevée et la PIO doit être surveillée étroitement. En cas daugmentations de la PIO ne répondant pas aux traitements ou interventions hypotonisants, limplant ILUVIEN peut être retiré par vitrectomie.
Les données concernant leffet de lacétonide de fluocinolone dans lil après une vitrectomie sont limitées. La clairance du médicament serait probablement accélérée après une vitrectomie, mais les concentrations à létat déquilibre ne devraient pas être modifiées. Cela peut diminuer la durée daction de limplant.
Dans les études FAME, 24 % des patients du groupe de traitement simulé ont reçu à un moment donné des médicaments anticoagulants ou antiplaquettaires versus 27 % des patients traités par ILUVIEN. Lincidence dhémorragies conjonctivales a été légèrement supérieure chez les patients traités par ILUVIEN de façon concomitante ou dans les 30 jours suivant larrêt des médicaments anticoagulants ou antiplaquettaires par rapport aux patients recevant le traitement simulé (0,5 % et 2,7 % respectivement dans les groupes traitement simulé et ILUVIEN). Le seul autre événement rapporté à une incidence plus élevée chez les patients traités par ILUVIEN a été une complication de la chirurgie oculaire (0 % et 0,3 % respectivement dans les groupes traitement simulé et ILUVIEN).
Il existe un risque de migration de limplant dans la chambre antérieure, en particulier chez les patients présentant des anomalies de la capsule postérieure, telles que des ruptures capsulaires. Cela doit être pris en compte lors de lexamen des patients se plaignant de troubles visuels après le traitement.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude dinteraction na été réalisée.
Il n'existe pas de données sur l'utilisation de lacétonide de fluocinolone administré par voie intravitréenne chez la femme enceinte. Les études effectuées chez lanimal sont insuffisantes pour permettre de conclure sur la toxicité sur la reproduction de lacétonide de fluocinolone administré par voie intravitréenne (voir rubrique 5.3). Bien que lacétonide de fluocinolone soit indétectable dans la circulation générale après un traitement intraoculaire local, la fluocinolone est néanmoins un corticoïde puissant et même de très faibles niveaux dexposition systémique peuvent présenter un certain risque pour le ftus en développement. Par mesure de précaution, il est préférable déviter lutilisation dILUVIEN pendant la grossesse.
Allaitement
Lacétonide de fluocinolone administré par voie systémique est excrété dans le lait maternel. Même si lexposition systémique de la femme qui allaite à lacétonide de fluocinolone administré par voie intravitréenne devrait être très faible, une décision doit être prise soit dinterrompre lallaitement soit de sabstenir du traitement avec ILUVIEN en prenant en compte le bénéfice de lallaitement pour lenfant au regard du bénéfice du traitement pour la femme.
Fertilité
Il nexiste pas de données concernant leffet dILUVIEN sur la fertilité. Cependant, des effets sur la fertilité masculine ou féminine sont peu probables dans la mesure où lexposition systémique à lacétonide de fluocinolone est très faible après administration par voie intravitréenne.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Lacétonide de fluocinolone administré par voie intravitréenne a été évalué chez 768 patients (375 dans le groupe 0,2 µg/jour/ILUVIEN ; 393 dans le groupe 0,5 µg/jour) présentant un dème maculaire diabétique dans les études cliniques FAME. Les effets indésirables les plus fréquents ont été : une chirurgie de la cataracte, une cataracte et une augmentation de la pression intraoculaire.
Dans les études de phase III, 38,4 % des patients traités par ILUVIEN ont eu besoin dun traitement hypotonisant et 4,8 % dune intervention chirurgicale pour réduire la PIO. Lutilisation dun traitement hypotonisant a été similaire chez les patients qui avaient reçu deux traitements ou plus par ILUVIEN.
Deux cas dendophtalmie ont été rapportés chez des patients traités par ILUVIEN dans les études de phase III, soit un taux dincidence de 0,2 % (2 cas/1 022 injections).
Bien que la majorité des patients des études cliniques FAME nait reçu quun seul implant (voir rubrique 5.1), les implications pour la tolérance à long terme de la rétention dans lil de limplant non bioérodable ne sont pas connues. Dans les études cliniques FAME, les données à 3 ans montrent que les événements tels que cataracte, augmentation de la pression intraoculaire et corps flottants ont été un peu plus fréquents chez les patients ayant reçu 2 implants ou plus. Cela est considéré comme étant dû à laugmentation de lexposition au médicament plutôt quà un effet de limplant lui‑même. Dans les études précliniques, il na pas été mis en évidence daugmentation des problèmes de tolérance autres que des modifications du cristallin chez des lapins ayant reçu 2 à 4 implants en 24 mois. Limplant est composé de polyimide et il est très similaire à une haptique de lentille intraoculaire ; il est donc attendu quil reste inerte dans lil.
Liste des événements indésirables
Les effets indésirables ci‑dessous ont été jugés comme étant liés au traitement et sont présentés selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000). Au sein de chaque fréquence de groupe, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
|
Infections et infestations |
Peu fréquent : endophtalmie |
|
Affections du système nerveux |
Peu fréquent : céphalées |
|
Affections oculaires |
Très fréquent :cataracte1, augmentation de la pression intraoculaire2 Fréquent : glaucome3, douleur oculaire4, hémorragie vitréenne, hémorragie conjonctivale, vision trouble5, baisse de lacuité visuelle, corps flottants du vitré Peu fréquent : occlusion vasculaire rétinienne6, affection du nerf optique, maculopathie, atrophie optique, ulcère conjonctival, néovascularisation de liris, exsudats rétiniens, dégénérescence vitréenne, décollement vitréen, opacification capsulaire postérieure, adhérences de liris, hyperémie oculaire, amincissement de la sclérotique, écoulement oculaire, prurit oculaire. |
|
Lésions, intoxications et complications liées aux procédures |
Peu fréquent : expulsion de limplant, implant dans la ligne de vision, complication opératoire, douleur opératoire |
|
Actes médicaux et chirurgicaux |
Très fréquent : opération de la cataracte Fréquent : trabéculectomie, chirurgie de glaucome, vitrectomie, trabéculoplastie Peu fréquent : retrait de limplant expulsé hors de la sclérotique |
|
Troubles généraux et anomalies au site d'administration |
Peu fréquent : déplacement de dispositif |
1 Inclut les termes MedDRA pour cataracte (NOS - sans autre précision), cataracte sous‑capsulaire, cataracte nucléaire et cataracte diabétique.
2 Inclut les termes MedDRA pour augmentation de la pression intraoculaire et hypertension oculaire.
3 Inclut les termes MedDRA pour glaucome, glaucome à angle ouvert, glaucome limite, excavation du nerf optique et augmentation du rapport cup/disc (rapport largeur de lexcavation/largeur de la papille).
4 Inclut les termes MedDRA pour douleur oculaire, irritation oculaire et gêne oculaire.
5 Inclut les termes MedDRA pour vision trouble et diminution de lacuité visuelle.
6 Inclut les termes MedDRA pour occlusion de la veine rétinienne, occlusion de lartère rétinienne et occlusion vasculaire rétinienne.
Description deffets indésirables sélectionnés
Lutilisation de corticoïdes au long cours peut provoquer des cataractes et une augmentation de la pression intraoculaire. Les fréquences mentionnées ci‑dessous reflètent les données observées chez lensemble des patients inclus dans les études FAME. Les fréquences observées chez les patients présentant un OMD chronique nont pas été significativement différentes de celles rapportées dans la population globale.
Dans les études cliniques de phase III, lincidence de cataracte chez les patients phaques a été denviron 82 % dans le groupe traité par ILUVIEN et de 50 % dans le groupe recevant le traitement simulé. Une chirurgie de la cataracte a été nécessaire après 3 ans chez 80 % des patients phaques traités par ILUVIEN versus 27 % des patients recevant le traitement simulé ; chez la plupart des patients, la chirurgie a été nécessaire au bout de 21 mois. La cataracte sous‑capsulaire postérieure est le type le plus fréquent de cataracte cortisonique. Pour ce type de cataracte, lintervention est plus difficile et peut être associée à un risque plus élevé de complications chirurgicales.
Dans les études FAME, les patients ayant une PIO initiale > 21 mmHg étaient exclus. Lincidence daugmentation de la pression intraoculaire a été de 37 % et un traitement hypotonisant a été nécessaire chez 38 % des patients, la moitié dentre eux ayant besoin dau moins deux médicaments pour équilibrer la PIO. Lutilisation dun traitement hypotonisant a été comparable chez les patients ayant été traités à nouveau par un autre implant pendant létude. De plus, une intervention chirurgicale ou un traitement au laser pour contrôler la PIO a été nécessaire chez 5,6 % (21/375) des patients qui avaient reçu un implant (trabéculoplastie, 5 patients [1,3 %], trabéculectomie, 10 patients [2,7 %], cycloablation endoscopique, 2 patients [0,5 %], et autres interventions chirurgicales, 6 patients [1,6 %]).
Dans le sous groupe de patients ayant une PIO supérieure à la médiane au début de létude (≥ 15 mmHg), un traitement hypotonisant a été nécessaire chez 47 % dentre eux et le pourcentage dinterventions chirurgicales ou de traitements au laser a augmenté à 7,1 %. Dans ce sous groupe, 5 patients (2,2 %) ont été traités par trabéculoplastie, 7 patients (3,1 %) par trabéculectomie, 2 patients (0,9 %) par cycloablation endoscopique et 4 patients (1,8 %) par dautres types de chirurgie du glaucome.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.ansm.sante.fr.
Aucun cas de surdosage na été rapporté.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Anti‑inflammatoires, corticoïdes, non associés, code ATC : S01BA15
Les corticoïdes inhibent la réponse inflammatoire à différents agents déclenchants. Ils inhibent la formation ddème, le dépôt de fibrine, la dilation capillaire, la migration leucocytaire, la prolifération capillaire, la prolifération des fibroblastes, le dépôt de collagène et la formation de cicatrices fibreuses associés à linflammation.
Les corticoïdes agiraient par induction des protéines inhibitrices de la phospholipase A, appelées lipocortines. On suppose que ces protéines contrôlent la biosynthèse des médiateurs puissants de linflammation tels que les prostaglandines et les leucotriènes en inhibant la libération de leur précurseur commun, lacide arachidonique. Lacide arachidonique est libéré des phospholipides membranaires par la phospholipase A2. Il a également été démontré que les corticoïdes diminuent les taux de facteur de croissance de lendothélium vasculaire (VEGF), une protéine qui augmente la perméabilité vasculaire et provoque ldème.
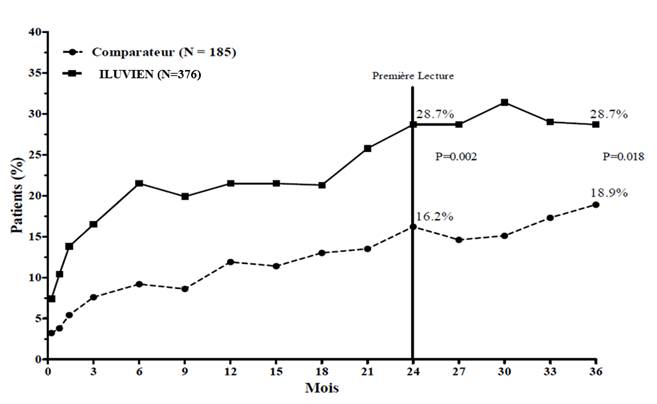
Lefficacité dILUVIEN a été évaluée dans deux études multicentriques randomisées en double aveugle, en groupes parallèles, menées chez des patients présentant un dème maculaire diabétique et ayant antérieurement reçu au moins un traitement par photocoagulation au laser. La durée de suivi était de trois ans pour chaque patient. Au total, 74,4 % des patients ont reçu 1 implant, 21,6 % ont reçu 2 implants, 3,5 % ont reçu 3 implants, 0,5 % ont reçu 4 implants et aucun patient na reçu plus de 4 implants. Dans les deux études, le critère principal dévaluation de lefficacité était le pourcentage de patients ayant obtenu un gain dacuité visuelle dau moins 15 lettres après 24 mois. Dans chacune de ces études, le critère principal a été atteint pour ILUVIEN (voir Figure 1 pour les résultats combinés du critère defficacité principal).
Figure 1. Pourcentage de patients ayant obtenu un gain ≥ 15 lettres par rapport au début de l'étude, études FAME combinées
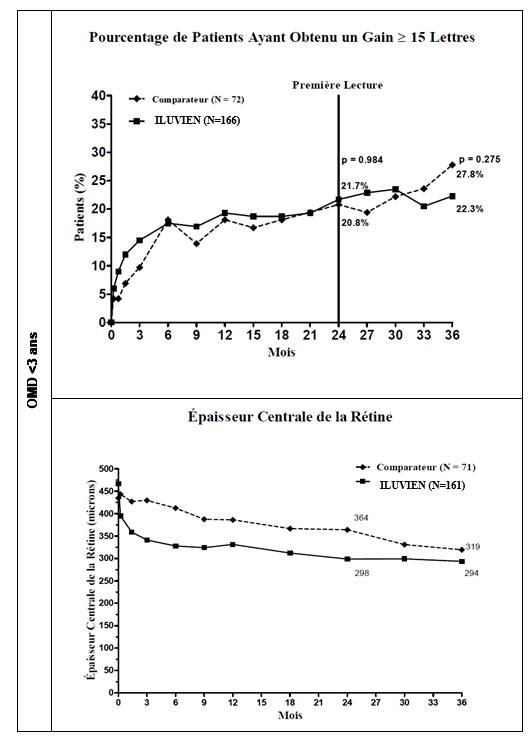
Lorsque lefficacité a été évaluée en fonction de la durée de la maladie, une réponse bénéfique significative a été observée chez les patients dont lancienneté de lOMD était supérieure à la médiane (≥ 3 ans), tandis que les patients chez lesquels la durée de lOMD était plus courte nont pas présenté de bénéfice supplémentaire par rapport au traitement comparateur en termes damélioration de lacuité visuelle (Figures 2 et 3). Ces données en sous‑groupes étayent lindication dans le traitement dOMD chronique (cest‑à‑dire dune durée dau moins 3 ans) qui figure à la rubrique 4.1.
Figure 2 : Comparaison des pourcentages de patients ayant obtenu un gain ≥ 15 lettres de la meilleure acuité visuelle corrigée (MAVC) par rapport à lentrée dans l'étude et variation moyenne de lépaisseur centrale de la rétine excessive, par sous‑groupe en fonction de la durée de lOMD ≥ 3 ans
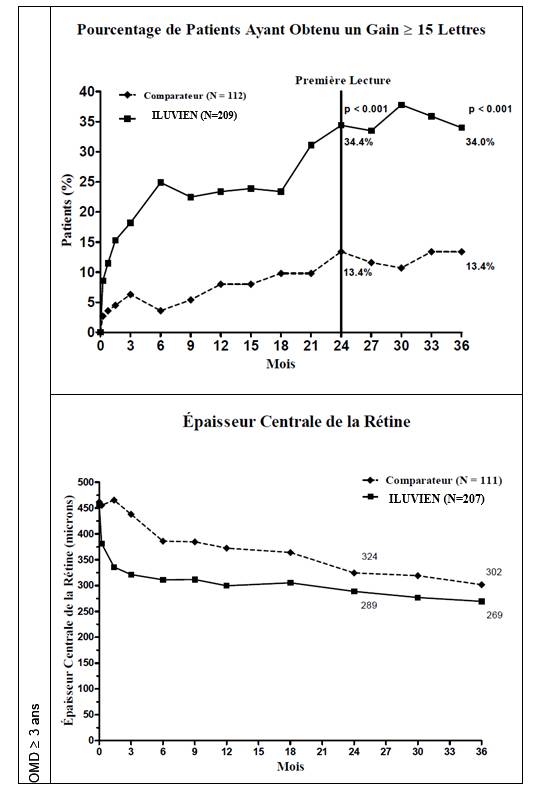
Figure 3 : Comparaison de la variation moyenne de lépaisseur centrale de la rétine excessive et pourcentage de patients ayant obtenu un gain ≥ 15 lettres de la MAVC par rapport à lentrée dans l'étude et variation moyenne de lépaisseur centrale de la rétine excessive, par sous‑groupe en fonction de la durée de lOMD < 3 ans
LAgence européenne des médicaments a accordé une dérogation à lobligation de soumettre les résultats détudes réalisées avec lacétonide de fluocinolone administré par voie intravitréenne dans tous les sous‑groupes de la population pédiatrique dans le traitement de ldème maculaire diabétique (voir rubrique 4.2 pour les informations concernant lusage pédiatrique).
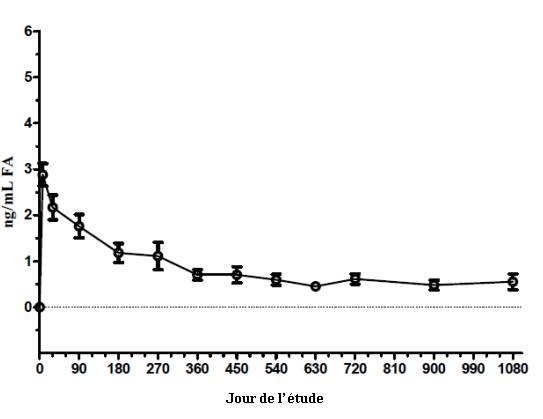
5.2. Propriétés pharmacocinétiques
Figure 4 : Concentrations de la FA dans lhumeur aqueuse humaine chez des sujets ayant reçu 1 implant ILUVIEN (Etude FAMOUS)
5.3. Données de sécurité préclinique
Des effets locaux (lésions dégénératives focales des fibres dans les régions polaire postérieure et corticale postérieure du cristallin) ont été observés chez le lapin à des doses dacétonide de fluocinolone administré par voie intravitréenne supérieures à la dose utilisée en clinique. Des effets locaux (fibrose rétinienne focale) ont également été observés chez des lapins traités avec le dispositif contenant lacétonide de fluocinolone ou le placebo. Cette formation de tissu fibreux na pas été observée en clinique chez lhomme et il est présumé quelle est due à des différences anatomiques entre lil de lapin et lil humain.
Alcool polyvinylique, tube en polyimide, adhésif siliconé.
Sans objet.
Après première ouverture de lopercule, utiliser immédiatement.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30°C. Ne pas mettre au réfrigérateur ou congeler.
La plaquette thermoformée scellée ne doit être ouverte quimmédiatement avant utilisation.
6.5. Nature et contenu de l'emballage extérieur
6.6. Précautions particulières délimination et de manipulation
Eliminer lapplicateur en toute sécurité dans un collecteur à aiguilles DASRI.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
ROYAL PAVILION
WELLESLEY ROAD
ALDERSHOT
HAMPSHIRE GU11 1PZ
ROYAUME-UNI
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Médicament à prescription réservée aux spécialistes en ophtalmologie.
Liste I.