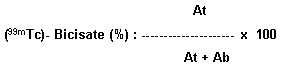
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 13/01/2017
NEUROLITE, trousse pour la préparation radiopharmaceutique : Injection de Bicisate de Technétium (99mTc).
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Flacon A : Poudre
Bicisate (dichlorhydrate de)................................................................................................... 0.90 mg
Flacon B : Solvant
Aucun principe actif nest présent.
Excipient(s) à effet notoire :
Le flacon A contient 0,04 mg de sodium (sous forme de 0,36 mg dédétate disodique).
Le flacon B contient 0,78 mg de sodium (sous forme de 4,1 mg de phosphate disodique heptahydraté et de 0,46 mg de dihydrogénophosphate de sodium monohydraté).
Après reconstitution de NEUROLITE avec une solution injectable, stérile et apyrogène, sans oxydant de pertechnétate (99mTc) de sodium, le complexe Tc-99m N,N'(1,2-ethylenediyl) bis-L-cysteine diéthyl ester (bicisate (99mTc)) est formé. (Le radio-isotope nest pas fourni avec le kit).
Pour la liste complète des excipients, voir rubrique 6.1.
Le kit contient deux flacons non radioactifs :
Flacon A: Poudre pour solution injectable
Flacon B: Solvant pour solution injectable.
4.1. Indications thérapeutiques
Ce médicament est à usage diagnostique uniquement.
La scintigraphie au bicisate (99mTc) est indiquée dans l'évaluation des anomalies de la perfusion régionale cérébrale chez les patients adultes atteints d'affections du système nerveux central.
4.2. Posologie et mode d'administration
Après marquage avec une solution injectable de pertechnétate (99mTc) de sodium, l'activité à administrer en injection intraveineuse chez un sujet moyen (70 kg) est de 740 MBq.
La scintigraphie devra être faite dans les 6 heures qui suivent l'administration.
Si nécessaire, une activité plus élevée jusqu'à 1700 MBq peut être injectée à condition que le patient soit capable d'uriner au moins toutes les 2 heures. La dose à injecter sera calculée au moyen dun système de mesure approprié de radioactivité avant dêtre administrée au patient. Il est recommandé de vérifier la pureté radiochimique avant ladministration au patient.
NEUROLITE nest pas recommandé pour les patients avec insuffisance rénale en raison du manque de données relatives à la tolérance et à lefficacité.
Population pédiatrique
NEUROLITE ne doit pas être utilisé chez lenfant en dessous de 18 ans compte tenu de labsence de données concernant sécurité et efficacité.
Mode dadministration
Pour les instructions relatives à la préparation et au contrôle de la pureté radiochimique des produits radiopharmaceutiques, voir rubrique 12.
Hypersensibilité à la substance active ou à lun des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Grossesse, voir rubrique 4.6
Pour chaque patient, lexposition à la radiation doit être justifiée par le bénéfice probable. Lactivité administrée doit, dans tous les cas, être aussi faible que raisonnablement possible pour obtenir les données de diagnostic nécessaires.
Insuffisance rénale
Une attention particulière du rapport bénéfice/risque est nécessaire chez ces patients dans la mesure où une augmentation prolongée de lexposition à la radiation est possible.
Préparation du patient
Les patients doivent être encouragés à augmenter la prise de liquides par voie orale sil ny a pas de contre-indication médicale à ceci, et à les évacuer aussi souvent que possible afin de réduire la radiation de la vessie.
Les produits radiopharmaceutiques ne doivent être reçus, utilisés et administrés que par des personnes habilitées, dans un environnement clinique autorisé. Leur réception, conservation, utilisation, transfert et élimination sont soumis aux réglementations et/ou aux licences appropriées prévue à l'article R 1333-24 du Code de la Santé Publique. Les produits radiopharmaceutiques seront préparés par lutilisateur dune manière qui répond à la fois aux exigences de sécurité de la radiation et de qualité pharmaceutique. Des précautions aseptiques appropriées seront prises, conformément aux exigences des Bonnes Pratiques de Fabrication des produits pharmaceutiques.
Le contenu des flacons est uniquement prévu pour la préparation du bicisate de technétium (99mTc) et ne doit pas être administré directement aux patients sans avoir subi auparavant la procédure de marquage.
Comme tout radiotraceur, le bicisate (99mTc) doit être manipulé avec précaution, des mesures de sécurité appropriées devant être appliquées pour minimiser l'exposition aux radiations du personnel hospitalier et des patients.
Une hydratation adéquate et une miction fréquente sont nécessaires afin de diminuer lirradiation de la vessie.
Pour chaque patient, lexposition à une radiation ionisante doit être justifiée sur une base de bénéfice probable. Lactivité administrée doit être telle que la dose de radiation en résultant soit aussi faible que raisonnablement possible en tenant compte de la nécessité dobtenir le diagnostic ou le résultat thérapeutique attendu.
Les produits radiopharmaceutiques doivent être préparés par lutilisateur de façon à satisfaire à la fois aux normes de radioprotection et de qualité pharmaceutique. Les précautions appropriées dasepsie doivent être prises afin de satisfaire aux exigences des Bonnes Pratiques de Fabrication pharmaceutique.
Si une hypersensibilité ou des réactions anaphylactoïdes se manifestent, ladministration du médicament sera immédiatement arrêtée et un traitement intraveineux introduit, le cas échéant. Pour permettre dagir rapidement en cas durgence, les médicaments et léquipement tels un dispositif dinsertion de tube endotrachéen et un ventilateur doivent être immédiatement disponibles.
Ce produit contient moins d1 mmol de sodium (23 mg) par dose, cest-à-dire quil est pratiquement «sans sodium».
Le débit sanguin cérébral pourrait être sous-estimé.
Les précautions en ce qui concerne les risques environnementaux figurent dans la rubrique 6.6.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction médicamenteuse na été décrite à ce jour.
4.6. Fertilité, grossesse et allaitement
Aucune étude de reproduction animale n'a été réalisée avec le bicisate (99mTc). De même qu'aucune étude n'a été effectuée chez la femme enceinte.
Quand il est nécessaire d'administrer des produits radioactifs à la femme susceptible de procréer, des informations doivent être obtenues quant à une grossesse. Toute femme ayant un retard de règles doit être présumée enceinte jusqu'à preuve du contraire. Quand il existe un doute, il est important que l'exposition aux radiations soit minimale compte tenu de l'information clinique souhaitée. On envisagera d'autres examens ne mettant pas en jeu de radiations ionisantes.
L'utilisation de radionucléides chez la femme enceinte implique également des doses de radiation pour le ftus. Seuls les examens indispensables seront effectués pendant la grossesse, quand le bénéfice attendu est supérieur aux risques encourus par la mère et le ftus.
Avant administration de produit radioactif à la femme allaitante, il faut envisager la possibilité de reporter jusqu'au sevrage et si le meilleur choix du produit radiopharmaceutique a été fait, compte tenu de sa sécrétion dans le lait maternel. Si l'administration est jugée nécessaire, l'allaitement doit être suspendu pendant les 12 heures qui suivent l'administration et le lait produit pendant cette période doit être éliminé. L'allaitement pourra être repris lorsque l'activité présente dans le lait ne risque pas d'entraîner une dose d'irradiation de l'enfant supérieure à 1mSv.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables rapportés suite à l'utilisation de NEUROLITE dans des études (1022 patients) étaient généralement d'une intensité légère à modérée et transitoires.
Au cours du programme d'essais cliniques, 5,9% des patients ont présenté un effet indésirable. Les effets indésirables les plus fréquemment rapportés sont des maux de tête (1,0%) et une agitation (0,5%).
La fréquence des effets indésirables listés ci-dessous est définie par la convention suivante:
Très fréquent (≥1/10); fréquent (≥1/100, <1/10); peu fréquent (≥1/1000, <1/100); rare (≥1/10000, <1/1000); très rare (<1/10000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Affections du système nerveux: |
Fréquent: maux de tête Peu fréquent: agitation, convulsions, parosmie (se manifestant par une légère odeur aromatique et transitoire), somnolence, hallucinations, anxiété, sensations vertigineuse |
|
Affections cardiaques: |
Peu fréquent: angor, insuffisance cardiaque |
|
Affections vasculaires: |
Peu fréquent: syncopes, hypertension |
|
Affections respiratoires, thoraciques et médiastinales: |
Peu fréquent: apnée, cyanose |
|
Affections gastro-intestinales: |
Peu fréquent: constipation, nausées, dyspepsie, diarrhée |
|
Affections de la peau et du tissu sous-cutané: |
Peu fréquent: éruption transitoire |
|
Troubles musculo-squelettiques et systémiques: |
Peu fréquent: douleurs dorsale |
|
Troubles généraux et anomalies au site d'administration: |
Peu fréquent: malaise |
|
Affections du système immunitaire :
|
Rare : réactions dhypersensibilité (exprimées sous forme de prurit, érythème, urticaire, nausées, gonflement du visage ou des lèvres, hyperémie oculaire, hypotension). Rare : réactions anaphylactiques de légères à graves (sous forme ddème, de gonflement des lèvres, dhypotension) |
L'exposition aux radiations ionisantes peut induire lapparition dun cancer et potentialiser le développement de malformations congénitales. Étant donné que la plupart des examens à visée diagnostique pratiqués en médecine nucléaire se font avec des doses à faible radiation inférieure à 20 mSv le taux de probabilité de ces effets indésirables semble faible. La dose efficace pour un adulte (70 kg) est de 13,6 mSv quand lactivité maximale recommandée de 1700 MBq est administrée.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Dans le cas dun surdosage de ladministration de radiation par Neurolite, la dose absorbée par le patient doit être réduite si possible en augmentant lélimination du radionucléide du corps au moyen de mictions et de défécations fréquentes. Il peut savérer utile destimer la dose effectivement délivrée.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Diagnostic radiopharmaceutique pour images du SNC.
Code ATC: V09AA02.
En raison des faibles concentrations administrées, on ne sattend pas à des effets pharmacodynamiques du bicisate (99mTc).
5.2. Propriétés pharmacocinétiques
Le bicisate (99mTc) peut exister sous quatre formes énantiomères différentes en fonction de la stéréochimie de la configuration du bicisate. Des études ont démontré que seul le dérivé L,L est retenu et métabolisé par l'encéphale de façon stéréo-sélective. A l'opposé, le complexe 99mTc dérivé de l'isomère D,D traverse la barrière hémato-encéphalique et est extrait par le cerveau, mais il n'est ni fixé, ni métabolisé dans des proportions appréciables. C'est pourquoi, seule la forme L,L est présente dans le NEUROLITE.
Des études conduites chez le volontaire sain ont montré que la fixation initiale du bicisate (99mTc) par le cerveau est de 4,8 à 6,5% de l'activité administrée, quelques minutes seulement après l'injection. La fixation et la rétention du bicisate (99mTc) par le cerveau sont suffisantes pour permettre d'obtenir une image en tomoscintigraphie par émission monophotonique (TEMP) immédiatement après administration. Le bicisate (99mTc) s'élimine très lentement du cerveau. Sa répartition dans l'encéphale reste stable pendant au moins six heures après l'injection. Cette répartition est comparable à celle observée avec le xénon-133, produit de référence pour l'étude du débit sanguin cérébral.
Le bicisate (99mTc) s'élimine rapidement du sang circulant. Une heure après administration, il reste dans le sang moins de 5% de l'activité injectée. Cinq minutes après injection, la plus grande partie de l'activité du sang veineux est due à la présence de métabolites. En moyenne, 74% de l'activité administrée sont excrétés dans les urines au cours des premières 24 heures suivant l'injection et jusqu'à 50% dans les deux premières heures.
Etant donné que la paroi vésicale est l'organe critique en terme d'irradiation et que l'élimination urinaire du bicisate (99mTc) est rapide, il est possible de réduire la dose délivrée en augmentant la fréquence des mictions. Ni le bicisate (99mTc), ni ses métabolites ne sont liés aux protéines plasmatiques.
Chez l'homme, comme avec d'autres radiotraceurs cérébraux marqués au technétium (99mTc), qu'une valeur de débit sanguin supérieure aux valeurs physiologiques normales pouvait entraîner une sous-estimation du débit relatif avec le bicisate (99mTc).
Aucune étude de pharmacocinétique du bicisate (99mTc) n'a été réalisée sur des patients présentant des troubles neurologiques. Toutefois, la distribution intracérébrale d'une dose unique de 99mTc-bicisate a été évaluée chez des patients avec le temps, à laide de tomoscintigraphies TEMP répétées réalisées par deux investigateurs, le Docteur Vidabaek et le Docteur Moretti. Les images de bicisate (99mTc) obtenues ont démontré la même distribution intracérébrale globale que dans la première étude de scintigraphie. Ceci semble indiquer que, comme chez les sujets neurologiques normaux, la distribution du bicisate (99mTc) reste stable avec le temps.
5.3. Données de sécurité préclinique
Les effets sur la reproduction et le pouvoir tératogène du bicisate (99mTc) n'ont fait l'objet d'aucune étude chez l'animal.
Il n'y a pas eu non plus d'études prolongées chez l'animal destinées à évaluer le pouvoir carcinogène ou à rechercher un effet du bicisate (99mTc) sur la fertilité chez le mâle ou la femelle.
Les résultats des tests mesurant les critères primaires defficacité indiquent que NEUROLITE n'est pas génotoxique in vitro et que son principe actif, le dichlorhydrate de bicisate (Bicisate2HCl), n'est pas génotoxique in vivo. Ces résultats soutiennent la conclusion selon laquelle NEUROLITE ne présente aucun aucun risque mutagène dans les conditions d'utilisation prévues.
Chlorure stanneux dihydraté
Édétate disodique
Mannitol
Acide chlorhydrique (pour ajustement du pH)
Flacon B :
Phosphate disodique heptahydraté
Dihydrogénophosphate de sodium monohydraté
Eau pour solution injectable
Il ne faut pas utiliser de solution injectable de pertechnétate (99mTc) de sodium contenant des oxydants.
Afin de préserver la stabilité du complexe technetié, les préparations de (99mTc) ne doivent pas être associées à d'autres préparations ou composants.
Durée de conservation de la trousse avant la reconstitution : 18 mois.
La durée de stabilité chimique et physique de la solution injectable reconstituée, en cours dusage, est de 8 heures à une température inférieure à 25°C. Ne pas mettre au réfrigérateur. Ne pas congeler.
6.4. Précautions particulières de conservation
Avant la reconstitution, les constituants du kit ne sont pas radioactifs. Toutefois, après avoir ajouté la solution injectable de pertechnétate (99mTc) de sodium, Ph. Eur., une protection adéquate de la préparation finale doit être assurée.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacons de verre borosilicate de 5 ml, Type I, (un flacon A et un flacon B), obturé avec un bouchon en halobutyle recouvert d'une capsule en aluminium sertie.
6.6. Précautions particulières délimination et de manipulation
Ladministration de médicaments radioactifs présente des risques pour lentourage du patient en raison de lirradiation externe ou de la contamination par les urines, les vomissements, etc. Par conséquent, il faut prendre des mesures de protection contre les radiations, conformément aux réglementations nationales.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
Festival House
39 Oxford Street
Newbury
Berkshire RG14 1JG
ROYAUME-UNI
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 558 375 6 5 : 25 mg en flacon (verre) et 1 mL de solution tampon en flacon (verre); boîte de 1.
· 34009 558 376 2 6 : 25 mg en flacon (verre) et 1 mL de solution tampon en flacon; boîte de 5.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Caractéristiques physiques du radionucléide utilisé pour le marquage:
Le technétium (99mTc) est produit au moyen un générateur (99Mo/99mTc). Il se désintègre avec une émission de radiation gamma avec une énergie moyenne de 141 keV et une demie-vie de 6,02 heures de technétium (99Tc) qui, en vue de sa longue demi-vie de 2,13 x 105 années, qui peut être considérée comme stable.
Les doses de radiation prévues pour les organes et les tissus dun patient de poids moyen (70 kg), après une injection intraveineuse de bicisate (99mTc), figurent dans le tableau ci-dessous :
Equivalent de dose absorbée par activité administrée [mGy/MBq]
|
Organe |
Equivalent de dose (mGy/MBq) |
|
|
Dose de radiation de 1700 MBq (miction 2,0 h après) |
Dose de radiation de 740 MBq (miction 4,8 h après) |
|
|
Cerveau Paroi de la vésicule biliaire Paroi de la partie inférieure du côlon Intestin grêle Paroi de la partie supérieure du côlon Reins Foie Poumons Ovaires Moelle rouge Surface des os Testicules Thyroïde Paroi vésicale Corps entier |
9,4 43 22 16 27 12 9,0 3,4 9,2 4,1 5,8 3,7 6,0 51 4,1 |
2,8 18,5 11,1 7,4 12,5 5,4 4,0 1,4 5,9 2,0 2,8 2,6 2,6 54,0 2,1 |
La dose efficace pour le bicisate (99mTc) est de 13,6 mSv à la dose maximale de 1700 MBq et des intervalles de miction de 2 heures. A la dose de 740 MBq et une miction après 4,8 heures, la dose efficace est de 7,7 mSv1.
(1)Dosimétrie calculée selon CIRP 60.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Ladministration de produits radiopharmaceutiques présente des risques pour lentourage du patient en raison de lirradiation externe ou de la contamination par les urines, les vomissements, etc. Par conséquent, il faut prendre des mesures de protection contre les radiations, conformément aux réglementations nationales.
Préparation de la trousse de NEUROLITE
La préparation du bicisate (99mTc) à partir de la trousse doit être réalisée de façon aseptique selon les étapes suivantes:
· Avant d'ajouter la solution injectable de pertechnétate (99mTc) de sodium au flacon B (flacon contenant la solution tampon), noter l'activité estimée, ainsi que la date et l'heure de la préparation, à l'endroit prévu à cet effet sur l'étiquette du flacon. Détacher ensuite une étiquette portant le symbole de radioactivité et la fixer autour du col du flacon.
· Porter des gants imperméables pendant la préparation. Enlever la rondelle en plastique de chacun des deux flacons et désinfecter la surface des bouchons à l'alcool.
· Placer le flacon B dans un conteneur blindé approprié portant une étiquette destiné à indiquer la date, l'heure de la préparation, le volume et l'activité.
· Utiliser une seringue blindée stérile pour ajouter au flacon B, de façon aseptique, 3,70 GBq (100 mCi) dans environ 2 mL de solution injectable, stérile et apyrogène, de pertechnétate (99mTc) de sodium sans oxydant. Sans retirer l'aiguille, enlever un volume d'air équivalent afin de maintenir la pression dans le flacon.
· A l'aide d'une seringue stérile, injecter rapidement 3 mL de chlorure de sodium (solution injectable, 0,9% NaCl, sans bactériostatique) dans le flacon A (le flacon contenant le lyophilisat) pour en dissoudre le contenu. Sans enlever l'aiguille, soustraire un volume d'air équivalent afin de rétablir dans le flacon la pression atmosphérique. Agiter le flacon pendant quelques secondes.
· A l'aide d'une autre seringue stérile, transférer immédiatement (dans les 30 secondes) 1,0 mL du flacon A au flacon B. Jeter immédiatement le flacon A.
· Mélanger le contenu du flacon B pendant quelques secondes, puis laisser reposer le mélange à température ambiante pendant trente (30) minutes.
· Avant injection, examiner le contenu du flacon B pour vérifier l'absence de particules et de changement de couleur. Si on observe des particules et/ou une variation de la coloration, NE PAS UTILISER
· Mesurer la radioactivité du flacon B au moyen d'un système de mesure approprié. Sur l'étiquette spécifique du conteneur blindé, noter la concentration de technétium (99mTc), le volume total, l'heure et la date de la mesure, la date de la péremption et le numéro du lot. Fixer cette étiquette sur le conteneur.
· Conserver le flacon contenant le bicisate (99mTc) à température inférieure à 25°C jusqu'à utilisation; le produit doit alors être prélevé de façon aseptique. Ne pas réfrigérer ni congeler. Le flacon ne contient aucun conservateur.
· Les déchets doivent être traités selon la réglementation nationale concernant les matériaux radioactifs.
Note: il est recommandé de suivre le mode opératoire ci-dessus pour reconstituer le produit.
Le produit doit être utilisé dans les 8 heures qui suivent la reconstitution.
Détermination de la pureté radiochimique par chromatographie en couche mince (CCM).
La vérification de la qualité du marquage doit être effectuée selon le mode opératoire décrit ci-dessous:
Matériaux pour réaliser la chromatographie en couche mince (CCM)
· Plaques de gel de silice Baker-Flex IB-F, 2,5 x 7,5 cm, Baker #2/4463/03 ou équivalent
· Solvant: acétate d'éthyle pour CLHP
· Activimètre ou compteur gamma pour mesurer la radioactivité
· Petite cuve de chromatographie
· Seringue et flacons blindés, en fonction des besoins.
Procédure par CCM
Déterminer la pureté radiochimique de la solution finale par chromatographie en couche mince (CCM) au moyen de plaques de gel de silice Baker-Flex IB-F ou équivalent en utilisant de l'acétate d'éthyle comme solvant.
Procédure Avec de lacétate déthyle frais, verser une quantité suffisante dans la cuve pour obtenir une hauteur de solvant de 3 à 4 mm. Fermer hermétiquement la cuve avec du Parafilm® et attendre 15 à 40 minutes pour que le solvant séquilibre. Il est important de respecter la phase de pré-équilibration et de préserver lintégrité de « lespace de tête » dans la cuve de chromatographie, afin de garantir la reproductibilité des résultats de la CCM.
Note: l'acétate d'éthyle est irritant pour la peau et les muqueuses et doit être manipulé sous hotte aussi souvent que possible.
Tracer avec un crayon des traits peu appuyés sur la plaque de CCM, à deux (2), quatre et demi (4,5) et sept (7) centimètres du bas de la plaque. Déposer environ 5 μl de la solution à injecter au centre de la plaque sur le trait situé à 2 cm. Pour ce faire, on peut utiliser une seringue munie d'une aiguille de 0,4 mm ou 0,5 mm en tenant la seringue en position verticale et en attendant qu'une goutte se forme. Le diamètre de la goutte déposée ne doit pas être supérieur à 10 mm. Laisser sécher la goutte sans attendre plus que 5 à 10 minutes
Placer la plaque dans la cuve de CCM pré-équilibrée et laisser migrer le front du solvant jusqu'à la ligne tracée à 7 centimètres de haut (environ 15 minutes). Enlever la plaque et laisser sécher dans une zone bien ventilée.
Calcul
Couper la plaque de CCM le long du trait situé à 4,5 cm au moyen de ciseaux. Utiliser un activimètre ou un compteur gamma pour mesurer l'activité. La partie supérieure contient le bicisate (99mTc) et la partie inférieure contient toutes les impuretés radioactives.
Calculer la pureté radiochimique à l'aide de l'équation suivante:
où At = activité de la partie supérieure
Ab = activité de la partie inférieure
Critères
Le Rf du bicisate (99mTc) est de 0,9 + 0,1; le colloïde, le TcO4- et le (99mTc) EDTA ne migrent pas. Si la pureté radiochimique est inférieure à 90%, ne pas utiliser la trousse et jeter la préparation.
Liste I.
Médicament réservé à lusage hospitalier.
Les produits radiopharmaceutiques ne doivent être utilisés que par des personnes qualifiées. Ils ne peuvent être délivrés qu'à des praticiens ayant obtenu l'autorisation spéciale prévue à l'article R 1333-24 du code de la Santé Publique.