
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 22/05/2017
PROPESS 10 mg, système de diffusion vaginal
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour un système de diffusion vaginal.
Pour la liste complète des excipients, voir rubrique 6.1.
Système de diffusion vaginal.
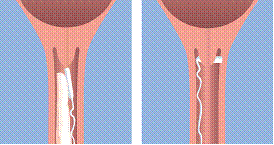
Propess se présente sous la forme dun système de diffusion vaginal polymérique, mince, plat, semi-transparent, de forme rectangulaire avec des coins arrondis contenu dans un système de retrait en polyester tissé.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Le système de diffusion vaginal doit être inséré assez haut dans le cul-de-sac postérieur du vagin
Le système de diffusion vaginal doit être retiré après 24 heures, que la maturation cervicale soit achevée ou non.
Après le retrait du système de diffusion vaginal, il est recommandé de respecter un intervalle de temps d'au moins 30 minutes avant l'utilisation d'ocytociques.
Population pédiatrique
La sécurité et lefficacité de Propess chez les jeunes filles enceintes âgées de moins de 18 ans nont pas été établies.
Aucune donnée nest disponible.
Mode dadministration
Propess doit être conservé dans le sachet et doit être sorti du congélateur immédiatement avant utilisation. Aucune décongélation nest nécessaire avant utilisation.
Le sachet présente un trait repère sur un côté. Lemballage doit être ouvert en suivant ce trait repère tout le long de lextrémité supérieure du sachet. Les ciseaux ou objets coupants qui pourraient endommager le dispositif de retrait ne doivent pas être utilisés.
L'ouverture située sur l'une des faces du système de retrait est prévue afin de permettre au fabricant de placer, en cours de fabrication, le système de diffusion vaginal dans le système de retrait.
Le système de diffusion vaginal ne devra en aucun cas être retiré du système de retrait.
Le système de diffusion vaginal doit être introduit haut dans le cul-de-sac postérieur du vagin. De petites quantités de lubrifiants hydrosolubles peuvent être utilisées si nécessaire afin de faciliter l'insertion du système.
Une fois le système de diffusion vaginal inséré, le ruban de retrait pourra être coupé avec des ciseaux, en s'assurant que la longueur de ruban soit suffisante pour permettre le retrait du système.
Aucune tentative ne doit être faite pour introduire le bout du ruban dans le vagin, le retrait du système serait alors plus difficile.
Après insertion, la patiente doit être allongée pendant 20 à 30 minutes.
Comme la PGE2 est diffusée en continu pendant 24 heures, il est important de surveiller à intervalles fréquents et réguliers les contractions utérines ainsi que l'état du ftus.
Retrait
Le système de diffusion vaginal peut être retiré rapidement et facilement en tirant doucement sur le ruban de retrait.
Lorsque le système de diffusion vaginal est retiré du vagin, il aura atteint 2 à 3 fois sa taille d'origine et sera flexible.
Dans certains cas, le système doit être retiré immédiatement du vagin (Voir section 4.4 Mises en gardes spéciales et précautions demploi).
PROPESS NE DOIT PAS ETRE UTILISE, ou LAISSE EN PLACE dans les cas suivants :
· Hypersensibilité à la substance active ou à lun des excipients mentionnés à la rubrique 6.1
· lorsque le travail a commencé ;
· lorsque des ocytociques et/ou dautres médicaments inducteurs du travail sont administrés ;
· lorsque des médicaments anti-inflammatoires non stéroïdiens y compris laspirine sont utilisés (leur administration doit être interrompue avant ladministration de PGE2) ;
· lorsque la survenue de contractions utérines fortes et prolongées n'est pas souhaitable, notamment en cas :
o d'antécédent de chirurgie utérine majeure, par exemple une césarienne, une myomectomie (voir rubrique 4.4 et 4.8);
o de disproportion fto-pelvienne;
o d'anomalie de présentation ftale;
o de souffrance ftale suspectée ou confirmée ;
o d'antécédent de chirurgie majeure (autre que des biopsies ou des abrasions cervicales) ou de rupture du col de lutérus ;
· antécédent récent de pathologie inflammatoire pelvienne, à moins qu'un traitement adéquat n'ait été instauré;
· placenta praevia ou saignement vaginal inexpliqué pendant la grossesse.
· dantécédent de plus de 3 accouchements à terme. (aucune étude chez les femmes ayant accouché à terme plus de 3 fois na été réalisés).
4.4. Mises en garde spéciales et précautions d'emploi
Létat du col de lutérus doit être évalué avec soin avant toute administration de Propess.
Propess ne doit être utilisé que sous surveillance ftale et utérine régulière, dans un établissement hospitalier disposant du matériel adéquat.
L'administration de médicaments anti-inflammatoires non-stéroïdiens y compris l'aspirine, doit être interrompue avant l'administration de PGE2.
Il est impératif de placer le système dans le cul-de-sac vaginal postérieur et de prendre garde à ne pas le positionner en intra-cervical extra-amniotique, ce qui exposerait à une hypertonie utérine sévère.
Après insertion du système de diffusion vaginal, on surveillera de manière stricte et régulière :
- la vitalité ftale (rythme cardiaque ftal),
- lactivité utérine (intensité, amplitude, fréquence des contractions utérines),
- létat du col.
Le système doit être retiré immédiatement du vagin lorsque la maturation du col est considérée comme complète ou dans lune des situations décrites ci-dessous:
- dès le début du travail. Dans le contexte du déclenchement du travail avec PROPESS, le début du travail est défini par la présence de contractions utérines régulières et douloureuses survenant toutes les 3 minutes, quelles que soient les modifications du col.
Deux points importants sont à noter :
· dès que des contractions régulières et douloureuses sont constatées sous Propess, ces contractions ne diminueront pas si le système reste en place, la PGE2 continuant à être libérée ;
· dès que des contractions utérines régulières et douloureuses sont constatées, le système de diffusion vaginal sera retiré quel que soit laspect du col de lutérus afin déviter tout risque dhypertonie utérine. En effet, certaines patientes, en particulier les multipares, peuvent présenter des contractions régulières et douloureuses sans modification apparente du col de lutérus. Leffacement et la dilatation du col de lutérus risquent de ne pas se produire tant que lactivité utérine naura pas débuté.
- en cas de contractions utérines prolongées ou excessives : dans ce cas il y a un risque dhypertonie utérine pouvant aller jusquà la rupture utérine,
- en cas de rupture spontanée ou provoquée des membranes,
- en cas de signe de souffrance ftale,
- en cas dévénements indésirables systémiques de la PGE2 survenant chez la mère, tels que nausées, vomissements, hypotension artérielle ou tachycardie,
- au moins 30 minutes avant dinstaurer une perfusion intraveineuse docytocine.
La réadministration de Propess nest pas recommandée car les effets dune seconde insertion nont pas été étudiés.
Quelques cas de rupture utérine ont été rapportés en association avec lutilisation de Propess principalement chez les patientes présentant une contre-indication au produit (voir rubrique 4.3). Par conséquent, Propess ne devrait pas être administré chez les patientes ayant subi précédemment une césarienne ou une chirurgie utérine entraînant un risque potentiel de rupture utérine et des complications obstétricales associées.
Utiliser le produit avec précaution en cas :
· dantécédent dhypertonie utérine,
· de glaucome,
· dasthme,
· de grossesse multiple (aucune étude na été réalisée dans ce cas),
· lorsquil y a eu rupture des membranes,
Les données dutilisation de Propess chez les patientes ayant une rupture des membranes sont limitées. Par conséquent, Propess doit être utilisé avec prudence chez ces patientes. Etant donné que la libération de dinoprostone du système peut être affectée par la présence du liquide amniotique, lactivité utérine ainsi que létat ftal doivent être particulièrement suivis.
Les effets de ce médicament nont pas été étudiés chez les patientes présentant des pathologies pouvant altérer le métabolisme ou lexcrétion de la PGE2 (maladies pulmonaires, hépatiques ou rénales). Dans ce cas, lutilisation de PGE2 n'est pas recommandée.
Les femmes âgées de plus de 35 ans, les femmes ayant rencontré des complications durant leur grossesse tels quun diabète gestationnel, une hypertension artérielle ou une hypothyroïdie, ou dont lâge gestationnel de la grossesse est supérieur à 40 semaines ont un plus grand risque de développer une Coagulation Intravasculaire Disséminée (CIVD). Ce risque est dautant plus élevé chez celles pour lesquelles le travail a été déclenché (voir rubrique 4.8). Ainsi, la dinoprostone et locytocine devraient être utilisées avec précaution chez ces femmes. Dans la phase de post-partum qui suit immédiatement laccouchement, le médecin devrait porter une attention particulière à tous les signes cliniques qui pourraient évoquer une CIVD (ex fibrinolyse).
Le clinicien doit être conscient que lutilisation de dinoprostone, de même que celle dautres méthodes de déclenchement du travail, peut entraîner une altération de la surface déchange par décollement placentaire et une embolie consécutive du tissu antigénique pouvant provoquer, dans de rares circonstances, le développement dun syndrome anaphylactoïde de la grossesse (embolie amniotique).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude dinteraction na été réalisée.
Les prostaglandines potentialisent les effets de l'ocytocine. Par conséquent, Propess ne doit pas être utilisé en même temps que l'ocytocine.
L'administration de médicaments anti-inflammatoires non-stéroïdiens y compris l'aspirine, doit être interrompue avant l'administration de PGE2.
4.6. Fertilité, grossesse et allaitement
Grossesse
Propess est uniquement indiqué pour linduction de la maturation et/ou de la dilatation du col lorsque la grossesse arrive à terme (37 semaines daménorhée révolues) et quun déclenchement du travail est indiqué.
Propess nest pas indiqué durant dautres phases de la grossesse.
Les études effectuées chez l'animal ont mis en évidence un effet tératogène.
En clinique, il n'existe pas actuellement de données suffisamment pertinentes pour évaluer un éventuel effet malformatif. En fin de grossesse, l'analyse d'un nombre élevé de grossesses exposées lors du déclenchement de l'accouchement à terme, n'a apparemment révélé aucun effet ftotoxique particulier de la dinoprostone.
Toutefois, seules des études épidémiologiques permettraient de vérifier l'absence de risque.
Aucune étude na été ménée pour évaluer la quantité de dinoprostone retrouvée dans le colostrum ou le lait maternel après lutilisation de Propess.
La dinoprostone pourrait être excrétée dans le colostrum ou le lait maternel, mais la quantité et la durée devraient être très limitées et ne devraient pas empêcher lallaitement. Aucun effet de la dinoprostone sur les nouveau-nés allaités na été observé dans les essais cliniques.
L'allaitement est possible au décours de l'utilisation de ce médicament.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les événements indésirables le plus fréquemment reportés dans les essais cliniques defficacité versus placebo ou comparateurs actifs (N=1116) ont été les troubles du rythme cardiaque ftal (6,9%), les contractions utérines anormales (6,2%) et le travail anormal affectant le ftus (2,6%).
Le tableau ci-dessous présente les principaux effets indésirables répartis selon les classes par systèmes et organes (SOC) et la fréquence. En outre, les effets indésirables observés post-commercialisation sont mentionnés avec une fréquence indéterminée.
|
MedDRA Système Classe Organe |
Fréquent (≥ 1/100 and < 1/10) |
Peu fréquent (≥ 1/1000 and ≤ 1/100) |
Indéterminée |
||||
|
Affections hématologiques et du système lymphatique |
|
|
Coagulation intravasculaire disséminée |
||||
|
Affections du système immunitaire |
|
|
Réaction anaphylactique Hypersensibilité |
||||
|
Affections du système nerveux |
|
Céphalée |
|
||||
|
Affections cardiaques |
Troubles du rythme cardiaque ftal 1* |
|
|
||||
|
Affections vasculaires |
|
Hypotension |
|
||||
|
Affections respiratoires, thoraciques et médiastinales |
|
|
|
||||
|
Affections gastro-intestinales |
|
|
Douleur abdominale Nausées, vomissements, diarhée |
||||
|
Affections hépatobiliaires |
|
Hyperbilirubinémie néonatale |
|
||||
|
Affections de la peau et du tissus sous-cutané |
|
Prurit |
|
||||
|
Affections gravidiques, puerpérales et périnatales |
Travail anormal affectant le ftus 2* Contractions utérines anormales 4* Méconium dans le liquide amniotique |
Hémorragie du post partum, Décollement prématuré du placenta, Score dApgar bas Arrêt du travail Chorioamniotite Atonie utérine |
Syndrome anaphylactoïde de la grossesse (embolie amniotique) Syndrome de détresse ftal 3* |
||||
|
Affections des organes de reproduction et du sein |
|
Sensation de brulure vulvovaginale |
dème génital |
||||
|
Troubles généraux et anomalies au site d'administration |
|
Troubles fébriles |
|
||||
|
Lésions, intoxications et complications liées aux procédures |
|
|
Rupture utérine |
1* : «Les troubles du rythme cardiaque ftal» rapportés dans les essais cliniques incluaient les items «rythme cardiaque ftal anormal», «bradycardie ftale», «tachycardie ftale», «absence inexpliquée de variabilité normale», «diminution du rythme cardiaque ftal», «décélération du rythme cardiaque ftal», «décélération précoce ou retardée», «décélération variable», «décélération prolongée».
2* : La dénomination «travail anormal affectant le ftus» rapporté dans les études cliniques, en tant que marqueur dun syndrome dhyperstimulation, incluait les « hypercinésies utérines » associées à des « décélérations retardées », des « bradycardies ftales » ou des « décélérations prolongées ».
3* : La dénomination « syndrome de détresse ftal » incluait les items « acidose ftale », « Enregistrement Cardiotocographique pathologique », « anomalie du rythme cardiaque ftal », « hypoxie intra utérine » ou « menace dasphyxie ». Cette dénomination en elle-même nest pas spécifique, a une faible valeur prédictive positive et est souvent associée avec un nouveau-né en bonne santé à la naissance.
4* : Les « contractions utérines anormales » incluaient les « hyperstimulations utérines » et l «hypertonie utérine».
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Un surdosage ou une hypersensibilité à la dipronostone peut provoquer une hypertonie utérine ou un retentissement ftal éventuel.
Dans ce cas, le système sera immédiatement retiré et un traitement symptomatique sera institué.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : PROSTAGLANDINES E2, code ATC : G02AD02
La prostaglandine E2 (PGE2) est une substance d'origine naturelle présente à de faibles concentrations dans la plupart des tissus de l'organisme. La prostaglandine E2 est une hormone à action locale.
Elle joue un rôle important dans le déroulement des modifications biochimiques et structurelles aboutissant à la maturation cervicale. Celle-ci nécessite la relaxation des fibres musculaires lisses du col de l'utérus qui se ramollit et se dilate pour permettre ainsi le passage du ftus dans la filière génito-pelvienne. Ce processus implique l'activation d'une enzyme: la collagénase qui est responsable de la dégradation du collagène.
Ainsi, l'administration locale de PGE2 au niveau du col de l'utérus conduit à la maturation cervicale qui permet la poursuite et l'achèvement du travail.
5.2. Propriétés pharmacocinétiques
Il n'a pas été possible d'établir de corrélation entre la libération de PGE2 et les concentrations plasmatiques de son métabolite PGEm, ni de déterminer les contributions relatives de PGE2 endogène et exogène, sur les taux plasmatiques du métabolite PGEm.
La PEG2 est libérée en continu au niveau cervical à une concentration qui permet la maturation cervicale jusqu'à la dilatation complète du col.
La dose moyenne libérée par heure est d'environ 0,3 mg pendant une période de 24 heures chez les femmes ayant des membranes intactes. En cas de rupture des membranes, la dose moyenne libérée est plus élevée et plus variable.
Le clinicien peut retirer le système de diffusion vaginal s'il décide que l'administration de PGE2 n'est plus nécessaire.
5.3. Données de sécurité préclinique
Les polymères d'hydrogel et de polyester sont des composants inertes, bien tolérés localement.
La toxicité sur la reproduction, la génotoxicité et la carcinogénèse des polymères n'ont pas été étudiées, mais l'exposition systémique paraît négligeable.
Hydrogel polymérique: macrogol 8000, dicyclohexyl-methane-4,4-diisocyanate, hexanetriol.
3 ans
6.4. Précautions particulières de conservation
A conserver dans le sachet non ouvert au congélateur entre -10°C et -20°C.
6.5. Nature et contenu de l'emballage extérieur
Chaque système de diffusion vaginal est contenu dans un système de retrait en polyester tissé et est conditionné individuellement dans un sachet de film thermosoudé en aluminium/polyéthylène. Boîte de cinq systèmes.
6.6. Précautions particulières délimination et de manipulation
Le système de diffusion vaginal doit être conservé dans le sachet et doit être sorti du congélateur immédiatement avant utilisation. Aucune décongélation nest nécessaire avant utilisation.
Le système dans son intégralité sera éliminé comme tout déchet médical.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
7, rue Jean-Baptiste Clément
94250 Gentilly
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
Date de dernier renouvellement : 03/02/2009 (illimité)
10. DATE DE MISE A JOUR DU TEXTE
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament réservé à lusage hospitalier