RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 10/08/2018
BELKYRA 10 mg/ml, solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Acide désoxycholique............................................................................................................ 10 mg
pour 1 mL de solution injectable
Chaque flacon contient 20 mg d'acide désoxycholique dans 2 ml de solution injectable.
Excipient(s) à effet notoire :
Chaque ml contient 184 µmol (ou 4,23 mg) de sodium provenant du chlorure de sodium, de l'hydroxyde de sodium et du phosphate disodique anhydre.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution limpide, incolore, sans particule visible.
Le pH de la solution est ajusté à 8,3 à l'aide d'acide chlorhydrique ou d'hydroxyde de sodium. La solution a une tonicité compatible avec celle des liquides et tissus biologiques avec une osmolalité de 300 mOsm/kg.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Le volume total injecté et le nombre de séances de traitement doivent être déterminés en fonction de la répartition de la graisse sous-mentonnière et des objectifs de traitement spécifiques à chaque patient.
Injectez 0,2 ml de solution de BELKYRA (soit 2 mg d'acide désoxycholique) par site d'injection, les injections devant être espacées d1 cm. La dose maximale de 10 ml de solution de BELKYRA (soit 100 mg d'acide désoxycholique correspondent à 50 injections) ne doit pas être dépassée au cours d'une même séance de traitement.
Le nombre maximal de séances de traitement est de 6. La plupart des patients présentent une amélioration après 2 à 4 séances de traitement.
Un intervalle minimum de 4 semaines entre deux séances de traitement doit être respecté.
Afin daméliorer le confort des patients pendant l'injection, des analgésiques oraux ou des AINS (anti-inflammatoires non-stéroïdiens), un anesthésique local topique et/ou injectable (par exemple lidocaïne), et/ou des poches de gel réfrigérant au niveau de la zone dinjection peuvent être utilisés, à lappréciation du médecin.
Populations particulières
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire (voir rubrique 5.2).
Insuffisance hépatique
Aucun ajustement posologique n'est nécessaire (voir rubrique 5.2).
Patients âgés (65 ans et plus)
Aucun ajustement posologique n'est nécessaire. La prudence est de rigueur chez les patients âgés (voir rubrique 4.4).
Population pédiatrique
Il nexiste pas dutilisation justifiée de BELKYRA dans la population pédiatrique.
Mode dadministration
BELKYRA doit être administré par voie sous-cutanée stricte.
BELKYRA doit être administré uniquement par des médecins ayant les qualifications adéquates, l'expérience du traitement et les connaissances nécessaires de l'anatomie sous-mentonnière. Lutilisation sûre et efficace de BELKYRA dépend dune sélection pertinente des patients, ce qui implique une connaissance de leurs antécédents médicaux notamment des interventions chirurgicales antérieures ayant pu modifier l'anatomie superficielle de la région cervicale. Une attention particulière doit être apportée lors de lutilisation de BELKYRA chez les patients présentant un relâchement excessif de la peau, des bandes platysmales proéminentes ou dautres particularités car chez ces patients, une diminution de la graisse sous-mentonnière pourrait aboutir à un résultat non souhaité.
Chaque flacon de BELKYRA ne doit être utilisé que pour une seule séance d'injection(s) par patient et lexcédent de produit non utilisé doit être éliminé conformément à la réglementation en vigueur.
BELKYRA est fourni en flacons à usage unique et prêts à l'emploi. Remuez délicatement le flacon en le retournant plusieurs fois avant de l'utiliser. Ne pas diluer.
Insérez l'aiguille perpendiculairement à la peau pour les injections de BELKYRA.
La position de l'aiguille par rapport à la mâchoire est très importante car cela réduit le risque de lésion du nerf marginal mandibulaire, une branche motrice du nerf facial. Une lésion de ce nerf se traduit par un sourire asymétrique dû à la paralysie du muscle abaisseur de la lèvre.
Pour éviter une lésion du nerf marginal mandibulaire :
· Ne pas injecter au-dessus du bord inférieur de la mâchoire.
· Ne pas injecter dans une région définie par une ligne de 1 à 1,5 cm sous le bord inférieur (allant de l'angle de la mâchoire jusqu'au menton).
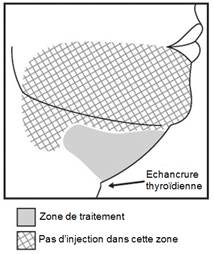
· Injecter BELKYRA uniquement dans la zone cible de traitement de la graisse sous-mentonnière (voir figures 1 et 3).
Figure 1. Évitez la région du nerf marginal mandibulaire
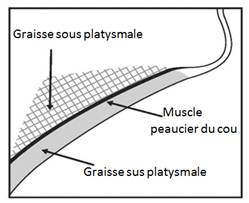
Ne pas injecter dans le muscle peaucier du cou (platysma). Avant chaque séance de traitement, palper la zone sous-mentonnière pour s'assurer de la présence d'une quantité suffisante de graisse sous-mentonnière et pour identifier la graisse sous-cutanée présente entre le derme et le muscle peaucier du cou (graisse sus platysmale (pré-platysmale)) située dans la zone cible du traitement (figure 2).
Figure 2. Vue sagittale de la zone du muscle peaucier
Tracer le contour de la zone cible du traitement avec un stylo chirurgical puis appliquer une grille d'injection dont chaque cellule mesure 1 cm2 afin de marquer lemplacement des sites d'injection (figures 2 et 3).
Figure 3. Zone de traitement et schéma d'injection
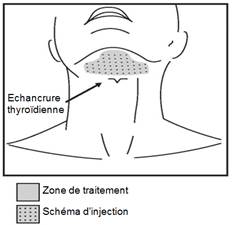
Ne pas injecter BELKYRA en dehors de la zone définie.
Il est nécessaire de vérifier visuellement la solution destinée à l'injection avant son utilisation. Seule une solution limpide, incolore et sans particule visible doit être utilisée.
· Hypersensibilité à lacide désoxycholique ou à lun des excipients mentionnés à la rubrique 6.1.
· Infection au niveau des sites dinjection prévus.
4.4. Mises en garde spéciales et précautions d'emploi
Administration par voie sous-cutanée stricte.
Injections dans ou à proximité de zones sensibles
Ne pas injecter dans une zone située à une distance de 1 à 1,5 cm de structures anatomiques sensibles.
BELKYRA ne doit pas être injecté dans ou à proximité de la branche marginale mandibulaire du nerf facial afin d'éviter une neurapraxie motrice, qui se manifeste par un sourire asymétrique ou une faiblesse des muscles du visage. Au cours des essais cliniques, les cas de lésions nerveuses étaient transitoires et se sont tous résolus.
Une attention particulière est nécessaire afin déviter toute injection intradermique ou intramusculaire accidentelle. BELKYRA doit être injecté à mi-profondeur du tissu adipeux sous-cutané sus platysmal (pré-platysmal) dans la zone sous-mentonnière. Ne pas retirer l'aiguille de la graisse sous-cutanée au cours de l'injection, car cela pourrait augmenter le risque de diffusion intradermique et conduire à d'éventuelles ulcérations cutanées.
Évitez toute injection dans les glandes salivaires, la glande thyroïde, les ganglions lymphatiques et les muscles.
Lefficacité et la sécurité demploi de BELKYRA en dehors de la graisse sous-mentonnière ou à des doses supérieures aux doses recommandées n'ont pas été établies. BELKYRA ne doit pas être utilisé chez les patients obèses (IMC ≥ 30) ni chez les patients présentant une dysmorphophobie
Antécédents de traitements ou de pathologies au niveau ou à proximité de la zone de traitement
Avant les injections de BELKYRA, les patients doivent être examinés pour rechercher d'autres causes potentielles de convexité sous-mentonnière (par exemple une hypertrophie de la thyroïde ou une lymphadénopathie cervicale).
Une attention particulière est nécessaire lors de ladministration de BELKYRA en cas d'inflammation ou d'induration au niveau des sites d'injection prévus ou chez les patients présentant des symptômes de dysphagie.
BELKYRA doit être administré avec précaution chez les patients ayant des antécédents de traitement chirurgical ou esthétique dans la zone sous-mentonnière. Des modifications dans l'anatomie/les repères anatomiques ou la présence de tissu cicatriciel dans cette zone peuvent présenter des risques en termes de sécurité pour ladministration de BELKYRA ou pour lobtention du résultat souhaité.
Patients âgés
Les études cliniques de BELKYRA n'ont pas inclus un nombre suffisant de patients âgés de plus de 65 ans pour déterminer s'ils répondent différemment en comparaison aux patients plus jeunes. Par conséquent, des précautions doivent être prises chez ces patients.
Patients suivant un régime pauvre en sodium
Ce médicament contient 184 µmol/ml (ou 4,23 mg/ml) de sodium. À prendre en compte chez les patients qui suivent un régime pauvre en sodium.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude dinteraction na été réalisée avec BELKYRA.
4.6. Fertilité, grossesse et allaitement
Grossesse
Des études sur la reproduction ont été réalisées sur des rats et des lapins, avec des expositions jusqu'à 1,8 fois (rat) et 12 fois (lapin) la dose maximale recommandée chez lHomme. Les études réalisées chez lanimal nont pas mis en évidence deffets délétères directs ou indirects sur la reproduction, cependant labsence de lobe moyen du poumon a été rapportée chez les lapins au cours de l'étude sur la toxicité embryonnaire et ftale (voir rubrique 5.3) sans pouvoir conclure sur le rôle de BELKYRA.
Il nexiste pas détude appropriée et contrôlée chez les femmes enceintes. Par mesure de précaution, il est préférable d'éviter l'utilisation de BELKYRA pendant la grossesse.
Il n'existe pas de données sur lexcrétion de l'acide désoxycholique dans le lait maternel, ni sur les effets du médicament chez le nourrisson allaité et sur la production de lait maternel. En labsence détudes conduites chez des femmes qui allaitent, des précautions sont nécessaires lors de ladministration de BELKYRA chez une femme qui allaite.
Fertilité
Il nexiste pas de données cliniques sur la fertilité.
BELKYRA n'a pas modifié les capacités générales de reproduction ni la fertilité chez les rats mâles et femelles à des doses allant jusqu'à 50 mg/kg, ce qui correspond respectivement à environ 5 et 3 fois la dose maximale recommandée chez lHomme (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Aucune étude n'a été réalisée sur laptitude à conduire des véhicules et à utiliser des machines.
Les données indiquées dans le tableau ci-dessous correspondent aux effets indésirables rapportés chez les patients traités par BELKYRA au cours des études cliniques destinées à évaluer BELKYRA pour le traitement de la graisse sous-mentonnière.
Les effets indésirables sont présentés par systèmes dorganes selon la classification MedDRA. Ils sont listés par fréquence, en utilisant la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classe de système d'organes |
Fréquence |
Effet indésirable |
|
Affections du système nerveux |
Fréquent |
Céphalées |
|
Peu fréquent |
Dysgueusie |
|
|
Affections respiratoires, thoraciques et médiastinales |
Peu fréquent |
Dysphonie |
|
Affections gastro-intestinales |
Fréquents |
Dysphagie, nausées |
|
Affections de la peau et du tissu sous-cutané |
Fréquent |
Raideur de la peau |
|
Troubles généraux et anomalies au site d'administration |
Très fréquents |
Au site d'injection : douleur, dème, gonflement, insensibilité, nodule, hématome, paresthésie, induration, érythème, prurit. |
|
Fréquents |
Au site d'injection : hémorragie, gêne, sensation de chaleur, décoloration |
|
|
Peu fréquents |
Au site d'injection : alopécie, urticaire, ulcère, hypersensibilité. |
|
|
Lésions, intoxications et complications liées aux procédures |
Fréquent |
Lésion nerveuse au site d'injection |
Généralement, la plupart des effets indésirables se sont résolus dans lintervalle entre deux séances de traitement. Le tableau suivant présente les effets indésirables signalés comme persistant plus longtemps que lintervalle de 4 semaines entre deux séances de traitement, sur la base des résultats des quatre études de phase 3 (N= 758) chez les patients traités par BELKYRA.
|
Effets indésirables |
BELKYRA |
Temps moyen jusqu'à la résolutiona (intervalle) |
|
Lésions nerveuses au site dinjection |
3,6 % |
53 jours (1-334 jours) |
|
Induration au site dinjection |
23,4 % |
41 jours (1-292 jours) |
|
Nodule au site dinjection |
12,0 % |
48 jours (1-322 jours) |
|
Douleur au site dinjection |
74,1 % |
12 jours (1-333 jours) |
|
Symptômes sensoriels au site d'injection |
66,4 % |
46 jours (1-349 jours) |
|
Insensibilité au site dinjection |
61,6 % |
50 jours (1-349 jours) |
|
Paresthésie au site dinjection |
11,3 % |
27 jours (1-297 jours) |
|
Gonflement au site dinjection |
78,6 % |
15 jours (1-218 jours) |
|
Dysphagie |
1,5 % |
22 jours (1-142 jours) |
a:Concerne le groupe BELKYRA uniquement.
Dans les études cliniques, certaines réactions locales, telles quinduration, nodule, insensibilité, douleur et gonflement au site dinjection, ainsi que des lésions du nerf moteur au site dinjection, ont été signalées comme non résolues à la fin des études cliniques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Aucun cas de surdosage de BELKYRA n'a été rapporté chez lHomme.
L'injection de plus grands volumes ou la réduction de l'espacement entre les sites d'injection de BELKYRA peut augmenter le risque d'effets indésirables locaux. Les événements indésirables systémiques ou survenant en dehors de la zone de traitement ont été rares au cours des études cliniques avec des doses allant jusqu'à 200 mg.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : autres préparations dermatologiques, code ATC : D11AX24.
Mécanisme daction
L'acide désoxycholique présente des propriétés cytolytiques. Une fois injecté localement dans la graisse sous-mentonnière, l'acide désoxycholique détruit la membrane cellulaire des adipocytes. La destruction des adipocytes provoque une réaction tissulaire au cours de laquelle les macrophages sont attirés au niveau de la zone d'injection afin d'évacuer les débris cellulaires et les lipides, qui sont ensuite éliminés de façon naturelle. Ce processus est suivi par l'apparition de fibroblastes et dun épaississement notable des cloisons fibreuses, suggérant une augmentation du collagène total (c'est-à-dire une néocollagenèse).
Efficacité et sécurité clinique
Quatre études de phase 3 randomisées, multicentriques, en double aveugle, contrôlées versus placebo ont été conduites (2 études identiques conduites dans l'Union Européenne (UE) et 2 études identiques conduites en Amérique du Nord) pour évaluer BELKYRA dans le traitement dune convexité liée à un excès de graisse sous-mentonnière ainsi que le retentissement psychologique associé. Dans toutes les études, les critères dévaluation principaux ont été mesurés 12 semaines après la dernière injection. L'efficacité a été démontrée dans chaque étude de phase 3 sur son critère principal d'efficacité et chaque étude de phase 3 a montré une amélioration en termes de retentissement psychologique par rapport au placebo.
Les études cliniques ont inclus des adultes (âgés de 19 à 65 ans) présentant une convexité modérée à sévère liée à un excès de graisse sous-mentonnière (c'est-à-dire avec un score de 2 ou 3 sur une échelle de 5 points, où 0 = absente, 5 = extrême), selon l'évaluation de linvestigateur et du sujet. Les patients ont reçu jusqu'à 4 traitements au cours des études conduites dans l'UE, et jusqu'à 6 traitements au cours des études conduites en Amérique du Nord, avec BELKYRA (N = 757 pour les 4 études) ou le placebo (N = 746) avec des intervalles de 28 jours entre chaque traitement. Le traitement a été interrompu lorsque la réponse souhaitée a été obtenue. Le volume d'injection était de 0,2 ml de solution de BELKYRA par site d'injection espacé de 1 cm dans la graisse sous-mentonnière, ce qui, exprimé en dose par zone de traitement, correspond à 2 mg/cm2 d'acide désoxycholique. Pour chaque séance de traitement, un maximum de 100 mg d'acide désoxycholique (soit 10 ml de BELKYRA) était autorisé pour l'ensemble de la zone de traitement.
L'âge moyen des patients dans les études conduites dans l'UE était de 46 ans et l'IMC moyen de 26 kg/m2. La majorité des patients étaient des femmes (75 %) de type caucasien (94 %). A linclusion, 68 % des patients présentaient une graisse sous-mentonnière modérée et 32 % une graisse sous-mentonnière sévère, évaluée par linvestigateur. Pour les études conduites en Amérique du Nord, l'âge moyen était de 49 ans et l'IMC moyen de 29 kg/m2. La majorité des patients étaient des femmes (85 %) de type caucasien (87 %). A linclusion, 51 % des patients avaient une graisse sous-mentonnière modérée et 49 % une graisse sous-mentonnière sévère, évaluée par linvestigateur.
Les critères dévaluation principaux combinés dans les études cliniques européennes étaient les scores de graisse sous-mentonnière (Clinician-Reported SubMental Fat Rating Scale CR-SMFRS) selon linvestigateur et l'évaluation de la satisfaction par le patient lui-même (échelle d'auto-évaluation par le sujet : Subject Self Rating Scale (SSRS)). Les scores de graisse sous-mentonnière (Patient-Reported SubMental Fat Rating Scale PR-SMFRS) attribués par le patient ont également été évalués. Le retentissement psychologique de la graisse sous-mentonnière a été évalué à l'aide de plusieurs mesures, dont l'échelle d'apparence Derriford 24 (Derriford Appearance Scale DAS-24), le score de qualité de vie liée à l'image du corps (Body Image Quality of Life Inventory BIQLI) et l'échelle de retentissement de la présence de graisse sous-mentonnière évaluée par le patient (Patient-Reported SubMental Fat Impact Scale PR-SMFIS), un questionnaire de 6 critères (évaluant le bien-être, la gêne, la perception de soi, l'embarras, la sensation d'être plus âgé ou en surpoids). Des améliorations statistiquement significatives concernant la graisse sous-mentonnière évaluée par linvestigateur et le patient, la satisfaction du patient et la diminution du retentissement psychologique de la graisse sous-mentonnière ont été plus fréquemment observées dans le groupe traité par BELKYRA que dans le groupe ayant reçu le placebo (Tableau 1). Une réduction du volume de la graisse sous-mentonnière a été confirmée par des mesures effectuées à l'aide d'un adipomètre.
Dans les études conduites en Amérique du Nord, les critères principaux dévaluation combinés étaient basés sur des améliorations d'au moins 1 ou 2 points pour la convexité sous-mentonnière, des scores composites des évaluations selon linvestigateur (CR-SMFRS) et selon le patient (PR-SMFRS) évaluant la graisse sous-mentonnière 12 semaines après la dernière injection. Le retentissement psychologique de la graisse sous-mentonnière a été évalué à l'aide du même questionnaire de 6 critères utilisé dans les études conduites dans l'UE. De plus, les modifications du volume de graisse sous-mentonnière ont été évaluées dans un sous-groupe de patients (N = 449, essais poolés) en utilisant l'imagerie par résonance magnétique (IRM). La réduction du volume de graisse sous-mentonnière a été confirmée à la fois par les mesures par IRM et par adipomètre.
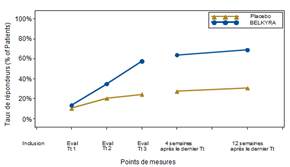
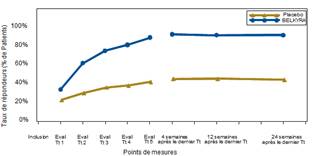
Le Tableau 1 ci-dessous présente les améliorations dau moins 1 point du score de graisse sous-mentonnière (CR-SMFRS) selon linvestigateur, les résultats de lévaluation de la satisfaction du patient (SSRS) et de l'amélioration du retentissement psychologique (PR-SMFIS), tels qu'ils ont été évalués dans les quatre études de phase 3. La figure 4 présente les taux de réponse sur la base des évaluations de la graisse sous-mentonnière par linvestigateur à chaque visite de l'étude.
Tableau 1 : Évaluation de la graisse sous-mentonnière* (GSM) par linvestigateur et le patient, de la satisfaction du patient et du retentissement psychologique à 12 semaines après le dernier traitement
|
|
Etudes conduites dans l'UE a |
Etudes conduites en Amérique du Nord b |
||
|
Critère |
BELKYRA (N = 243) |
Placebo (N = 238) |
BELKYRA (N = 514) |
Placebo (N = 508) |
|
Taux d'amélioration de 1 point de GSM* selon linvestigateur (CR-SMFRS) c |
63,8 % |
28,6 % |
78,5 % |
35,3 % |
|
Taux d'amélioration de 1 point de GSM* selon le patient (PR-SMFRS) c |
63,1 % |
34,3 % |
80,3 % |
38,1 % |
|
Taux de satisfaction du patient (SSRS) d |
65,4 % |
29 % |
69,1 % |
30,5 % |
|
Amélioration du retentissement psychologique (PR-SMFIS) en (moyenne des pourcentages par rapport à linclusion) e |
44,6 % |
18,0 % |
48,6 % |
17,3 % |
a Jusqu'à 4 séances de traitement autorisées
bJusqu'à 6 séances de traitement autorisées
cDiminution dau moins 1 point du score de la GSM* évaluée par linvestigateur (CR-SMFRS)12 semaines après la dernière injection.
dCorrespond à une évaluation de la satisfaction par le patient cotée « extrêmement satisfait(e) », « satisfait(e) » ou « légèrement satisfait(e) » sur l'échelle SSRS12 semaines après le dernier traitement.
e Amélioration moyenne en pourcentage par rapport à linclusion, obtenue en divisant la différence moyenne entre le score PR-SMFIS à la semaine 12 et linclusion, par le score moyen à linclusion.
Figure 4 : Taux de répondeurs basé sur le score de la graisse sous-mentonnière évalué par linvestigateur (CR-SMFRS) à chaque visite de l'étude ; données poolées à partir des études conduites dans l'UE (graphique de gauche) et en Amérique du Nord (graphique de droite)*
|
|
|
* p < 0,001 à tous les points de mesures, BELKYRA versus placebo
Bien que la majorité des patients ait présenté des réductions du volume de graisse sous-mentonnière, 90,0 % et 92 % respectivement des patients ayant participé aux études dans l'UE et en Amérique du Nord nont eu aucun changement (68,9 % et 70,5 %) ni amélioration (21,6 % et 22,9 %) des scores de relâchement de la peau 12 semaines après le dernier traitement, en comparaison avec linclusion.
La sécurité à long terme et le maintien de l'effet du traitement après administration de BELKYRA ont été évalués. Un sous-groupe de patients ayant présenté initialement une réponse au traitement par BELKYRA a été inclus dans des études de suivi où le maintien de leffet du traitement a été démontré jusquà plus de 5 ans.
Population pédiatrique
L'utilisation de BELKYRA n'est pas recommandée chez les sujets âgés de moins de 18 ans.
LAgence européenne des médicaments a accordé une dérogation à lobligation de soumettre les résultats détudes réalisées avec BELKYRA dans tous les sous-groupes de la population pédiatrique dans le traitement dune convexité modérée à sévère liée à un excès de graisse sous-mentonnière, lorsque la présence de graisse sous-mentonnière entraîne un retentissement psychologique chez le patient (voir rubrique 4.2 pour les informations concernant lusage pédiatrique).
5.2. Propriétés pharmacocinétiques
Absorption
L'acide désoxycholique provenant de BELKYRA est rapidement absorbé après injection sous-cutanée. Après injection de la dose maximale recommandée de BELKYRA pour une séance de traitement (100 mg), les concentrations plasmatiques maximales (Cmax moyenne) ont été observées avec un tmax médian de 6 minutes après l'injection. La valeur moyenne de Cmax était de 1036 ng/ml et était 2,3 fois supérieure aux valeurs moyennes de Cmax observées au cours d'une période de référence de 24 heures en l'absence d'injection de BELKYRA. A la dose maximale recommandée par traitement (100 mg), l'exposition à l'acide désoxycholique (ASC0-24) était moins de 2 fois supérieure à lexposition à l'acide désoxycholique endogène. LASC0-24 plasmatique a augmenté de manière proportionnelle à la dose jusqu'à 100 mg. Après le traitement, les concentrations plasmatiques dacide désoxycholique ont diminué pour revenir aux valeurs des concentrations de lacide désoxycholique endogène en 24 heures. Aucune accumulation n'est attendue avec la fréquence de traitement proposée.
Distribution
Le volume de distribution a été estimé à 193 L et est indépendant de la dose jusqu'à 100 mg. L'acide désoxycholique est fortement lié aux protéines plasmatiques (98 %).
Élimination
L'acide désoxycholique endogène est un produit du métabolisme du cholestérol et est excrété intact dans les selles. L'acide désoxycholique provenant de BELKYRA s'ajoute au pool d'acides biliaires endogènes et est excrété en même temps que l'acide désoxycholique endogène. L'acide désoxycholique est éliminé par les protéines de transport hépatiques, du sang vers la bile sans intervention significative du métabolisme.
L'acide désoxycholique n'est pas un inhibiteur in vitro des enzymes CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 et 3A4. Sur le plan clinique, lacide désoxycholique n'a pas entraîné l'induction du CYP1A, 2B6 et 3A.
L'acide désoxycholique n'est pas un inhibiteur in vitro des transporteurs BSEP, MRP2, MRP4, MDR1, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, OATP2B1 et ASBT. L'acide désoxycholique a inhibé le NTCP avec une CI50 de 2,14 µM in vitro.
Insuffisance rénale
BELKYRA n'a pas été étudié chez les patients présentant une insuffisance rénale. Les acides biliaires, y compris l'acide désoxycholique, sont excrétés dans l'urine en quantités négligeables ; il est donc peu probable que l'insuffisance rénale influence la pharmacocinétique de l'acide désoxycholique.
Insuffisance hépatique
BELKYRA n'a pas été étudié chez les patients présentant une insuffisance hépatique. Étant donné la fréquence d'administration discontinue, la faible dose administrée qui représente environ 3 % de l'ensemble des acides biliaires et la grande variabilité des concentrations endogènes de l'acide désoxycholique, il est peu probable que la pharmacocinétique de l'acide désoxycholique après injection de BELKYRA soit modifiée en cas d'insuffisance hépatique.
Patients âgés
Aucun ajustement de la posologie n'est nécessaire. La prudence est de rigueur chez les patients âgés (voir rubrique 4.4).
5.3. Données de sécurité préclinique
Carcinogénicité
Dans les études de toxicité en administration répétée de BELKYRA allant jusqu'à 6 mois chez le rat et 9 mois chez le chien, aucun élément évoquant une réponse pré-néoplasique locale ou généralisée suite à l'administration sous-cutanée de BELKYRA na été observé. Au cours de ces études, la dose clinique maximale fixée était dépassée de 2,5 à 12,5 fois (calculée en mg/site d'injection) et de 2 à 3 fois (selon une exposition systémique quantifiée) chez le rat et le chien respectivement. En outre, au lieu du rythme dadministration clinique maximal défini d'injections mensuelles jusqu'à 6 séances, BELKYRA a été administré au rythme de deux fois par mois, jusqu'à 13 fois la dose totale chez le rat et 20 fois la dose totale chez le chien. Aucune étude sur la carcinogénicité n'a été réalisée avec BELKYRA.
Génotoxicité
BELKYRA a été négatif dans une série de tests standards de toxicologie génétique in vitro (essai de mutation réverse sur des bactéries et essai d'aberration chromosomique) et in vivo (test du micronoyau).
Toxicité sur la reproduction et le développement
Une absence de lobe moyen du poumon a été rapportée chez les lapins au cours de l'étude sur la toxicité embryonnaire et ftale sans pouvoir conclure sur le rôle de BELKYRA. Le nombre de cas était significativement plus élevé dans le groupe à 30 mg/kg mais cette observation a été également mise en évidence à la concentration plus faible de 10 mg/kg. Cette dose était associée à une toxicité maternelle locale. La pertinence clinique de cette observation n'est pas établie.
Eau pour préparations injectables, chlorure de sodium, hydroxyde de sodium (pour dissolution et ajustement du pH), phosphate disodique anhydre, acide chlorhydrique (pour ajustement du pH).
30 mois.
Le produit doit être utilisé immédiatement après la perforation du bouchon du flacon.
En cas dutilisation non immédiate, les durées et conditions de conservation du flacon entamé relèvent de la responsabilité de lutilisateur.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
Pour les conditions de conservation du médicament après première ouverture, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Solution injectable en flacon (verre de type I), muni d'un bouchon (caoutchouc chlorobutyle) et dune bague de sécurité (aluminium) avec un couvercle à clapet (polypropylène).
Une boîte contient 4 flacons. Chaque flacon contient 2 ml de solution injectable.
6.6. Précautions particulières délimination et de manipulation
Chaque flacon est destiné à l'usage d'un seul patient.
Après utilisation, tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Préparer BELKYRA comme suit en vue de réaliser linjection :
1. Retirer le capuchon amovible du flacon et nettoyer le bouchon perforable du flacon à l'aide d'un antiseptique. Si le flacon, la bague de sécurité ou le capuchon amovible est endommagé(e), ne pas utiliser le flacon.
2. Fixer une aiguille stérile de gros calibre à une seringue stérile de 1 ml à usage unique.
3. Insérer laiguille stérile de gros calibre dans le bouchon du flacon et aspirer 1 ml de BELKYRA dans la seringue de 1 ml.
4. Remplacer laiguille de gros calibre par une aiguille de 30 G (ou plus petite) de 12,7 mm de longueur. Évacuer toute bulle d'air présente dans le corps de la seringue avant dinjecter le produit dans la graisse sous-cutanée.
5. Pour récupérer le contenu restant dans le flacon, répéter les étapes 3 et 4.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
ALLERGAN PHARMACEUTICALS INTERNATIONAL LTD
CLONSHAUGH INDUSTRIAL ESTATE
COOLOCK
17 DUBLIN
IRLANDE
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 300 731 9 3 : 2 ml de solution injectable en flacon (verre). Boîte de 4.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament réservé à lusage professionnel selon larticle R.5121-80 du code de la santé publique.
Prescription réservée aux spécialistes en chirurgie maxillo-faciale ou en chirurgie plastique, reconstructrice et esthétique ou en oto-rhino-laryngologie - chirurgie cervico-faciale.