RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 01/08/2018
OCTANATE 100 Ul/ml, poudre et solvant pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Octanate 100 Ul/ml contient 1000 UI de facteur VIII de coagulation humain par flacon.
Le produit contient approximativement 100 UI* par ml de facteur VIII de coagulation humain lorsquil est reconstitué avec 10 ml de solvant.
Produit à partir du plasma de donneurs humains.
Le produit contient approximativement ≤ 60 Ul/ml de facteur de Willebrand (vWF:RCo).
Excipient à effet notoire :
Sodium jusquà 1,75 mmol (40 mg) par dose
Concentration de sodium après reconstitution : 125 175 mmol/l
Pour la liste complète des excipients, voir rubrique 6.1.
*Lactivité (UI) est déterminée en utilisant le dosage chromogénique de la Pharmacopée Européenne. Lactivité spécifique moyenne dOctanate est ≥ 100 Ul/mg de protéine.
Poudre et solvant pour solution injectable.
La poudre est blanche ou jaune pâle sous forme dun solide friable.
Le solvant est un liquide limpide, incolore.
4.1. Indications thérapeutiques
Cette préparation ne contient pas de facteur de Willebrand en quantité pharmacologiquement active et par conséquent nest pas indiqué dans la maladie de Willebrand.
4.2. Posologie et mode d'administration
Posologie
La posologie et la durée du traitement de substitution dépendent de la sévérité du déficit en facteur VIII, de la localisation et de limportance de lépisode hémorragique ainsi que de létat clinique du patient.
Le nombre dunités de facteur VIII administrées est exprimé en Unités Internationales (UI), ramenées au standard actuel de lOMS pour les préparations de facteur VIII. Lactivité coagulante du facteur VIII dans le plasma est exprimée soit en pourcentage (par rapport au plasma humain normal) soit en Unités Internationales (par rapport à un standard international du facteur VIII plasmatique).
Traitement à la demande
Une Unité Internationale (UI) dactivité coagulante du facteur VIII correspond à la quantité de facteur VIII contenue dans un ml de plasma humain normal. Le calcul de la dose nécessaire de facteur VIII est basé sur le résultat empirique quune UI de facteur VIII par kg de poids corporel augmente lactivité coagulante du facteur VIII plasmatique de 1,5% -2% de lactivité normale. La posologie nécessaire est déterminée à laide de la formule suivante:
Unités nécessaires = poids corporel (kg) x augmentation souhaitée du taux de facteur VIII (%) (Ul/dl) x 0,5
La quantité à administrer et la fréquence dadministration doivent toujours être guidées par lefficacité clinique individuelle.
Dans le cas des événements hémorragique suivants, lactivité coagulante du facteur VIII ne doit pas chuter en dessous du taux dactivité coagulante plasmatique indiqué (en % de la normale) pendant la durée mentionnée.
Le tableau ci-dessous peut être utilisé comme guide pour les posologies lors dépisodes hémorragiques et de chirurgie:
|
Degré de lhémorragie/Type dintervention chirurgicale |
Taux de facteur FVIII nécessaire (%) |
Fréquence des doses (heures)/Durée de traitement (Jours) |
|
Hémorragie |
||
|
Début dhémarthrose, saignement musculaire ou buccal |
20 -40
|
Renouveler toutes les 12 à 24 heures sur au moins 1 jour, jusquà ce que lépisode hémorragique soit résolu comme indiqué par la douleur ou que la cicatrisation soit obtenue. |
|
Hémarthrose plus étendue, hémorragie musculaire ou hématome |
30 -60 |
Renouveler la perfusion toutes les 12 à 24 heures pendant 3 à 4 jours ou plus jusquà ce que la douleur et le handicap disparaissent. |
|
Hémorragies mettant en jeu le pronostic vital |
60 -100 |
Renouveler la perfusion toutes les 8 à 24 heures, jusquà disparition du risque vital. |
|
Chirurgie |
||
|
Mineure Dont extraction dentaire |
30-60 |
Toutes les 24 heures, au moins 1 jour, jusquà ce que la cicatrisation soit obtenue. |
|
Majeure |
80 -100 (pré et post opératoire) |
Renouveler la perfusion toutes les 8 à 24 heures, jusquà cicatrisation suffisante de la plaie, puis poursuivre le traitement pendant au moins 7 jours supplémentaires pour maintenir lactivité coagulante du FVIII entre 30% et 60%. |
Prophylaxie
Pour le traitement prophylactique à long terme des épisodes hémorragiques chez les patients atteints dhémophilie A sévère, les posologies sont de 20 à 40 UI de facteur VIII ·par kg de poids corporel, espacées de 2 à 3 jours. Dans certains cas, notamment chez les patients plus jeunes, des intervalles dadministration plus courts ou des posologies plus élevées peuvent être nécessaires.
Perfusion continue
Une analyse pharmacocinétique doit être effectuée avant une intervention chirurgicale pour obtenir une estimation de la clairance.
La vitesse initiale de perfusion peut être calculée de la manière suivante : clairance x concentration souhaitée à létat déquilibre = vitesse de perfusion (UI/kg/h).
La clairance doit être de nouveau calculée quotidiennement après les 24 premières heures de perfusion continue selon la formule ci-dessus.
Au cours du traitement, une détermination appropriée de facteur VIII est conseillée afin dévaluer la dose à administrer et la fréquence du renouvellement des perfusions. Dans le cas dinterventions chirurgicales majeures, un contrôle précis du traitement substitutif au moyen des tests de coagulation (activité plasmatique du facteur VIII) est indispensable. La réponse au traitement par le facteur VIII, les taux de récupération in vivoet les demi-vies observées peuvent varier selon les individus.
Patients précédemment non traités
Les données cliniques sur lutilisation dOctanate chez des patients précédemment non traités sont limitées. Les données actuellement disponibles sont décrites à la rubrique 5.1.
Population pédiatrique
Une étude clinique menée chez 15 patients âgés de 6 ans ou moins na pas démontré la nécessité dadapter la posologie chez les enfants.
Mode dadministration
Voie intraveineuse. Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Il est recommandé de ne pas administrer plus de 2 à 3 ml par minute.
Hypersensibilité à la substance active ou à lun des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Des réactions dhypersensibilité de type allergique sont possibles avec Octanate. Le produit contient des traces de protéines humaines autres que le facteur VIII. En cas dapparition de symptômes dhypersensibilité, il doit être conseillé aux patients darrêter immédiatement lutilisation du produit et de contacter leur médecin. Les patients doivent être informés des premiers signes dhypersensibilité, notamment: démangeaisons, urticaire généralisée, oppression thoracique, respiration sifflante, hypotension et anaphylaxie.
En cas de choc, le traitement médical standard relatif à létat de choc devra être instauré.
Inhibiteurs
L'apparition d'anticorps neutralisants (inhibiteurs) du facteur VIII est une complication connue du traitement des patients atteints d'hémophilie A. Ces inhibiteurs sont habituellement des immunoglobulines IgG dirigées contre lactivité coagulante du facteur VIII et sont mesurées en Unités Bethesda par ml de plasma par le test modifié. Le risque de développer des inhibiteurs est corrélé à la gravité de la maladie ainsi quà l'exposition au facteur VIII, ce risque étant le plus élevé au cours des 20 premiers jours d'exposition. Rarement, les inhibiteurs peuvent apparaître après les 100 premiers jours d'exposition.
Des cas de réapparition dinhibiteurs (faible titre) ont été observés après le changement dun facteur VIII pour un autre, chez des patients préalablement traités ayant plus de 100 jours dexposition et qui avaient des antécédents de développement dinhibiteur. Il est donc recommandé de surveiller attentivement tous les patients afin de détecter lapparition dun inhibiteur suite à un changement de produit.
La pertinence clinique de lapparition dinhibiteurs dépendra du titre dinhibiteurs ; un faible titre dinhibiteurs provisoire ou constant présente un risque de réponse clinique insuffisante moins élevé quun titre élevé dinhibiteurs.
De manière générale, tous les patients traités avec des produits de facteur VIII de coagulation doivent faire lobjet dune surveillance soigneuse pour détecter lapparition d'inhibiteurs par un suivi clinique et à l'aide de tests biologiques appropriés. Si le taux de facteur VIII plasmatique attendu nest pas atteint ou si lhémorragie nest pas contrôlée par une dose adéquate, un dosage doit être réalisé afin de rechercher la présence dun inhibiteur du facteur VIII. Chez les patients présentant un titre élevé dinhibiteur, le traitement en facteur VIII peut ne pas être efficace et dautres options thérapeutiques doivent être considérées. Le suivi de tels patients doit être effectué par des médecins expérimentés dans la prise en charge de lhémophilie et des inhibiteurs du facteur VIII.
Complications liées au cathéter
Si un dispositif daccès veineux central (DAVC) est nécessaire, un risque dinfections locales, de bactériémie ou de thrombose sur le site dinsertion du cathéter existe.
Agents infectieux transmissibles
Les mesures habituelles de prévention du risque de transmission dagents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, la recherche des marqueurs spécifiques dinfection sur chaque don et sur les mélanges de plasma ainsi que la mise en uvre dans le procédé de fabrication détapes efficaces pour linactivation/élimination virale. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission dagents infectieux ne peut pas être totalement exclu. Ceci sapplique également aux virus inconnus ou émergents ou autres types dagents infectieux.
Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de limmunodéficience humaine (VIH), le virus de lhépatite B (VHB) et de lhépatite C (VHC), et vis-à-vis du virus non-enveloppé de lhépatite A (VHA). Les mesures prises peuvent être defficacité limité vis-à-vis des virus non-enveloppés tels que le parvovirus B19. Linfection par le parvovirus B19 peut être sévère chez les femmes enceintes (infection ftal) et chez les personnes atteintes dun déficit immunitaire ou de certains types danémies.
Une vaccination appropriée (hépatite A et B) des patients recevant de façon régulière à répétée des préparations de facteur VIII plasmatique humain devra être envisagée.
Ce médicament contient jusquà 1,75 mmol de sodium (40 mg) par dose de 500 UI/flacon. À prendre en compte chez les patients suivant un régime contrôlé en sodium.
Population pédiatrique
Les mises en garde et précautions demploi indiquées concernent aussi bien les adultes que les enfants.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction des produits à base de facteur VIII humain avec dautres spécialités pharmaceutiques na été rapportée à ce jour.
4.6. Fertilité, grossesse et allaitement
Grossesse
Aucune étude sur la reproduction animale na été menée avec le facteur VIII. En raison de la rareté de lhémophilie A chez les femmes, lexpérience acquise lors de lutilisation de facteur VIII pendant la grossesse et lallaitement nest pas disponible. Aussi le facteur VIII ne doit être administré au cours de la grossesse et de lallaitement quen cas dindication absolue.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Une hypersensibilité ou des réactions allergiques (qui peuvent inclure angio-dème, sensation de brûlure et de piqûre au site dinjection, frissons, rougeurs, urticaire généralisée, céphalées, démangeaisons, hypotension, léthargie, nausées, agitation, tachycardie, oppression thoracique, fourmillements, vomissements, respiration sifflante) ont été rarement observées, et peuvent dans certaines cas évoluer vers une réaction anaphylactique sévère (voire un état de choc).
La fièvre a été observée à de rares occasions.
Des anticorps neutralisants (inhibiteurs) peuvent apparaître chez des patients atteints dhémophilie A traités avec le facteur VIII, y compris avec OCTANATE, voir rubrique 5.1. Une telle apparition se manifeste par une réponse clinique insuffisante. Dans ce cas, il est recommandé de contacter un centre spécialisé en hémophilie.
Pour des informations sur la sécurité concernant les agents transmissibles, voir la rubrique 4.4.
Liste des événements indésirables
Le tableau présenté ci-dessous est conforme à la classification par discipline médicale ou système organe classe (SOC) et au niveau de termes préférés MedDRA.
Les fréquences ont été évaluées selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1000 à < 1/100), rare (≥ 1/10 000 à < 1/1000), très rare (< 1/10 000), fréquence inconnue (ne peut être estimée à partir des données disponibles).
|
Norme MedDRA Classes de systèmes dorganes
|
Effets indésirables |
Fréquence des effets indésirables |
|
Affections du système immunitaire |
Hypersensibilité Choc anaphylactique |
Rare Très rare |
|
Troubles généraux et réactions au site dadministration |
Pyrexie |
Rare |
|
Affections hématologiques et du système lymphatique |
Inhibition du facteur VIII |
Peu fréquent (PPT)* Très fréquent (PUP)* |
|
Investigations |
Anticorps anti-facteur VIII positifs |
Rare |
* La fréquence est déterminée daprès des études sur des produits de facteur VIII menées auprès de patients atteints dhémophilie A sévère. PPT = patients précédemment traités, PUP = patients non traités précédemment (previously-untreated patients).
Population pédiatrique
La fréquence, le type et la sévérité des effets indésirables sont identiques chez les enfants et chez les adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
Aucun cas de surdosage na été rapporté.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmaco-thérapeutique : anti-hémorragiques : facteur VIII de coagulation sanguine.
Code ATC : B02BD02.
Le complexe facteur VIII/facteur von Willebrand se compose de deux molécules (FVIII et FvW) aux fonctions physiologiques différentes. Administré à un patient hémophile, le facteur VIII se lie au facteur Willebrand dans la circulation du patient.
Le facteur VIII activé agit comme cofacteur du facteur IX activé, accélérant la conversion du facteur X en facteur X activé. Le facteur X activé convertit la prothrombine en thrombine. La thrombine convertit ensuite le fibrinogène en fibrine, ce qui aboutit à la formation dun caillot.
Lhémophilie A est un déficit de la coagulation sanguine héréditaire et lié au sexe, dû à une diminution du taux de facteur VIII: C, provoquant des accidents hémorragiques profus au niveau des articulations, des muscles, ou des organes internes, soit spontanément ou résultant de traumatismes accidentels ou chirurgicaux. A laide dun traitement substitutif, les taux plasmatiques du facteur VIII sont augmentés, permettant ainsi une correction temporaire du déficit en ce facteur et une correction des tendances hémorragiques.
Patients précédemment non traités
Dans une étude clinique en cours chez des patients non préalablement traités (PUPs), 3 patients sur 39 (7,6 %) traités à la demande avec Octanate ont développé des inhibiteurs à un titre supérieur à 5 Unités Bethesda (UB). Un patient a développé des inhibiteurs avec un titre inférieur à 5 UB. Deux cas (5,1 %) étaient cliniquement significatifs ; les deux derniers patients ont développé des inhibiteurs qui ont disparu spontanément sans modification de la posologie dOctanate. Tous les inhibiteurs sont apparus avec un traitement à la demande et avant 50 jours dexposition.
35 PUPs avaient un taux basal dactivité de facteur VIII< 1 % et 4 PUPs avaient un taux basal de facteur VIII £ 2 %. Lors de lanalyse intermédiaire, le nombre de jours dexposition à Octanate était égal ou supérieur à 20 jours pour 34 patients, et égal ou supérieur à 50 jours pour 30 patients. Aucun inhibiteur na été détecté chez les patients traités en prophylaxie par Octanate. Au cours de létude, 12 PUPs ont subi 14 interventions chirurgicales. Lâge médian lors de la première exposition était de 7 mois (intervalle entre 3 jours et 67 mois). La médiane de jours dexposition était de 100 (intervalle entre 1 et 553).
Une étude clinique observationnelle visant à évaluer lefficacité dOctanate dans lInduction de Tolérance Immune (ITI) est en cours. Une analyse intermédiaire des 69 patients traités jusquà présent par Octanate dans le cadre dITI montre que 49 patients ont terminé létude. Chez les patients dont linhibiteur a été éliminé avec succès, la fréquence mensuelle des épisodes hémorragiques a été significativement réduite.
5.2. Propriétés pharmacocinétiques
Le facteur VIII de coagulation humain plasmatique (issu de la poudre) est un constituant normal du plasma humain et se comporte comme le facteur VIII endogène. Après administration, approximativement deux tiers à trois quarts du facteur VIII restent dans la circulation. Le niveau dactivité du facteur VIII obtenu dans le plasma devrait être entre 80% -120% de lactivité prédite du facteur VIII.
Distribution
Lactivité coagulante plasmatique du facteur VIII diminue suivant une courbe exponentielle décroissante en deux phases. Dans la phase initiale, la distribution entre le compartiment intravasculaire et les autres compartiments (fluides corporels) se fait avec une demi-vie délimination du plasma de 3 à 6 heures. Dans la phase suivante, plus lente (qui reflète probablement la consommation du facteur VIII), la demi-vie varie entre 8 et 20 heures, avec une moyenne de 12 heures. Ceci correspond à la vraie demi-vie biologique.
Pour Octanate, les résultats suivants ont été obtenus au cours de deux études pharmacocinétiques, avec respectivement 10 et 14 patients atteints dhémophilie A :
|
|
Récupération (% x UI-1 x kg) |
AUC*norm (% x h x UI-1 x kg) |
Demi-vie (h) |
MRT* (h) |
Clairance (ml x h-1 x kg) |
|
Etude 1, n=10 Moyenne ± SD* |
2,4 ± 0,36 |
45,5 ± 17,2 |
14,3 ± 4,01 |
19,6 ± 6,05 |
2,6 ± 1,21 |
|
Etude 2, n=14 Moyenne ± SD* |
2,4 ± 0,25 |
33,4 ± 8,50 |
12,6 ± 3,03 |
16,6 ± 3,73 |
3,2 ± 0,88 |
*SD : Déviation standard
*AUC : Aire sous la courbe
*MRT : Temps de résidence moyen
5.3. Données de sécurité préclinique
Chez lanimal, aucun effet toxique na été mis en évidence après administration de ces réactifs correspondant à des doses plusieurs fois multiples de la posologie humaine recommandée par kilogramme de poids corporel.
Aucun potentiel mutagène na été observé pour aucune des deux substances.
Citrate de sodium
Chlorure de sodium
Chlorure de calcium
Glycine
Solvant :
Eau pour préparations injectables
Ce médicament ne doit pas être mélangé avec dautres produits ou médicaments.
Seuls les dispositifs dinjection/perfusion fournis doivent être utilisés car un échec du traitement peut se produire comme résultant de ladsorption du facteur VIII de coagulation humain sur la surface interne de certains matériels dinjection/perfusion.
La solution reconstituée doit être utilisée immédiatement et en une seule fois.
6.4. Précautions particulières de conservation
A conserver au réfrigérateur (2°C - 8°C).
Ne pas congeler. Conserver les flacons dans lemballage extérieur à labri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Un emballage dOctanate contient :
Poudre en flacon (verre de type I), muni dun bouchon (caoutchouc chlorobutyle ou bromobutyle) et dun capuchon amovible.
Solvant en flacon (verre de type I), muni dun bouchon (caoutchouc chlorobutyle ou bromobutyle) et dun capuchon amovible.
Une seringue jetable, un nécessaire de transfert Mix2Vial, une aiguille dinjection et 2 compresses imbibées dalcool.
Les deux présentations disponibles diffèrent en termes de quantité de facteur VIII de coagulation humain/solvant:
Un flacon contient 1000 UI de facteur VIII de coagulation humain.
6.6. Précautions particulières délimination et de manipulation
Lire attentivement les instructions et les suivre scrupuleusement !
Ne pas utiliser Octanate après la date de péremption mentionnée sur l'étiquette.
Durant la procédure décrite ci-dessous, la stérilité doit être maintenue!
La solution dans la seringue doit être limpide ou légèrement nacrée. Ne pas injecter de solutions troubles ou ayant des dépôts.
Utiliser immédiatement la solution préparée afin de prévenir toute contamination microbienne.
Nutiliser que le nécessaire dinjection fourni. L'utilisation d'un autre équipement d'injection/perfusion peut induire des risques additionnels et un échec du traitement.
Instructions pour la préparation de la solution:
Ne pas utiliser le produit directement après la sortie du réfrigérateur. Amener le solvant et la poudre contenus dans les flacons fermés à température ambiante.
Retirer les capuchons amovibles des deux flacons et nettoyer les bouchons en caoutchouc avec lune des compresses imbibées d'alcool fournies.
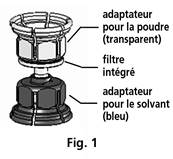
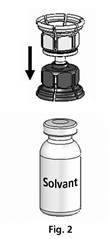
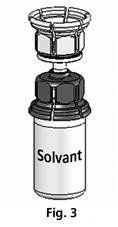
Le Mix2vial est représenté en Fig. 1. Placer le flacon de solvant sur une surface plane et le tenir fermement. Prendre le Mix2vial et le retourner. Placer la partie bleue du Mix2vial sur le dessus du flacon de solvant et appuyer fermement jusqu'à ce qu'il s'enclenche (Fig. 2 + 3).
|
Placer le flacon de poudre sur une surface plane et le tenir fermement. Prendre le flacon de solvant avec le Mix2vial fixé et le retourner. Placer la partie transparente sur le dessus du flacon de poudre et appuyer fermement jusqu'à ce qu'il s'enclenche (Fig. 4). Le solvant s'écoule automatiquement dans le flacon de poudre. |
|
|
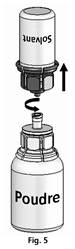
Les deux flacons toujours fixés, tourner doucement le flacon de poudre jusqu'à ce que le produit soit dissous. La dissolution est terminée en moins de 10 minutes à température ambiante. Il peut se produire une légère formation de mousse pendant la préparation. Dévisser le Mix2vial en deux parties (Fig. 5). La mousse va disparaître.
Eliminer le flacon de solvant vide avec la partie bleue du Mix2vial.
|
|
Instructions pour l'injection:
À titre de précaution, le pouls doit être mesuré avant et pendant l'injection. S'il se produit une forte augmentation de la fréquence cardiaque, réduire la vitesse d'injection ou interrompre l'administration pendant un court moment.
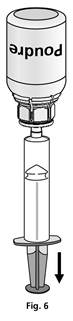
Fixer la seringue à la partie transparente du Mix2vial. Retourner le flacon et prélever la solution dans la seringue (Fig. 6).
La solution présente dans la seringue doit être limpide ou légèrement nacrée.
Dès que la solution a été transférée, tenir fermement le piston de la seringue (en la tenant tournée vers le bas) et retirer la seringue du Mix2vial (Fig. 7). Eliminer le Mix2vial et le flacon vide.

Nettoyer le site d'injection choisi avec lune des compresses imbibées d'alcool fournies.
Fixer l'aiguille pour injection fournie à la seringue.
Introduire l'aiguille pour injection dans la veine choisie. Si un garrot a été utilisé pour rendre la veine plus facile à voir, ce garrot doit être relâché avant de commencer à injecter Octanate.
Du sang ne doit pas pénétrer dans la seringue, en raison du risque de formation de caillots de fibrine.
Injecter la solution dans la veine lentement, pas plus vite que 2 à 3 ml par minute.
Si plus d'un flacon de Octanate poudre est utilisé pour un traitement, la même aiguille pour injection et la même seringue peuvent être réutilisés. Le Mix2vial est réservé à un usage unique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription initiale hospitalière de six mois (les établissements de transfusion sanguine autorisés à dispenser des médicaments dérivés du sang aux malades qui y sont traités, inclus).
La délivrance est réservée aux pharmacies à usage intérieur des établissements de santé ou aux établissements de transfusion sanguine pour les malades qui y sont traités.