RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 21/11/2018
ZEULIDE 3,75 mg, poudre et solvant pour suspension injectable à libération prolongée
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 3,75 mg dacétate de leuproréline (équivalent à 3,57 mg de leuproréline sous forme de base libre).
1 mL de suspension reconstituée contient 1,875 mg dacétate de leuproréline.
Excipients à effet notoire :
Chaque flacon contient entre 1,3 et 2,2 mg (<1 mmol) de sodium (sous forme de carmellose sodique).
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour suspension injectable à libération prolongée.
Poudre : poudre blanche à blanchâtre.
Solvant : solution limpide, incolore, exempt de particules visibles (pH 5,0 7,0).
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
La dose habituelle recommandée de ZEULIDE est de 3,75 mg présentés en une injection à libération prolongée sur un mois et administrés en une injection intramusculaire une fois par mois.
ZEULIDE 22,5 mg doit être administré sous la surveillance dun médecin ou dun professionnel de santé qui dispose de l'expertise appropriée de la surveillance de la réponse au traitement.
La dose de ZEULIDE 3,75 mg permettant la libération continue d'acétate de leuproréline pendant un mois est incorporée dans une formulation à effet retard.
La poudre lyophilisée doit être reconstituée et administrée en une unique injection intramusculaire chaque mois. Ladministration par voie intra-artérielle ou intraveineuse doit être évitée. Le flacon de poudre de microsphères de ZEULIDE 3,75 mg doit être reconstitué immédiatement avant ladministration du médicament par injection intramusculaire. Comme pour tout autre médicament administré par injection, il est nécessaire de changer régulièrement le point d'injection.
Le traitement par ZEULIDE 3,75 mg ne doit pas être interrompu même en cas de rémission ou damélioration.
La réponse au traitement par ZEULIDE 3,75 mg doit être contrôlée en mesurant régulièrement les taux sériques de testostérone et dantigène prostatique spécifique (PSA). Des études cliniques ont montré que les taux de testostérone augmentent pendant les 4 premiers jours de traitement chez la majorité des patients non orchidectomisés. Ils diminuent ensuite atteignant des taux de castration au bout de 3 à 4 semaines. Une fois atteints, ces taux de castration (définis par un taux de testostérone inférieur ou égal à 0,5 ng/mL) se maintiennent tout au long du traitement.
Chez les patients ayant une réponse sous-optimale, il est conseillé de confirmer que les taux sériques de testostérone ont atteint ou se maintiennent aux taux de castration. Des augmentations transitoires des taux de phosphatase acide surviennent parfois en début de traitement mais reviennent généralement à des valeurs normales ou proches de la normale dès la 4e semaine de traitement.
Durée du traitement
ZEULIDE 3,75 mg est administré sous forme dinjections intramusculaires mensuelles.
En règle générale, le traitement du cancer avancé de la prostate avec ZEULIDE 3,75 mg est un traitement à long terme ; le traitement ne doit pas être arrêté en cas de rémission ou d'amélioration.
Populations particulières
Population pédiatrique
La sécurité et lefficacité de ZEULIDE 3,75 mg dans la population pédiatrique nont pas été établies. Par conséquent, ZEULIDE 3,75 mg nest pas recommandé chez les enfants et les adolescents tant que des données de sécurité et defficacité ne sont pas disponibles.
Insuffisance rénale / hépatique
La pharmacocinétique de ZEULIDE 3,75 mg chez les patients atteints dinsuffisance hépatique ou rénale na pas été déterminée.
Population âgée
Dans lessai clinique portant sur ZEULIDE 3,75 mg, lâge moyen des sujets étudiés était de 71,0 ±9,02 ans. Par conséquent, les données figurant sur létiquetage concernant la pharmacocinétique, lefficacité et la sécurité de ZEULIDE 3,75 mg se réfèrent à cette population.
Mode dadministration
ZEULIDE 3,75 mg doit être administré uniquement par voie intramusculaire. Ne pas administrer par une autre voie. En cas dadministration sous-cutanée accidentelle, le patient doit être étroitement surveillé car aucune donnée nest disponible concernant ladministration ZEULIDE 3,75 mg par dautres voies que linjection intramusculaire. Pour les instructions sur la reconstitution du médicament avant administration, voir la rubrique 6.6.
Orchidectomie antérieure.
ZEULIDE 3,75 mg ne doit pas être utilisé seul chez les patients ayant un cancer de la prostate et présentant des signes de compression de la moelle épinière ou de métastases spinales.
ZEULIDE 3,75 mg nest pas indiqué chez la femme.
ZEULIDE 3,75 mg nest pas indiqué dans la population pédiatrique.
4.4. Mises en garde spéciales et précautions d'emploi
Lors des premières phases du traitement par ZEULIDE 3,75 mg, comme lors des traitements avec dautres agonistes de la GnRH, une augmentation transitoire des taux de testostérone peut se produire. Dans certains cas, ceci peut être associé à une « poussée » ou à une exacerbation de la croissance tumorale qui se traduit par une aggravation temporaire des symptômes du cancer de la prostate. Ces symptômes se dissipent généralement en poursuivant de traitement (voir rubrique 4.8). La « poussée » peut parfois se manifester par des symptômes systémiques ou neurologiques (par ex. douleurs osseuses, etc.). De plus, des cas datrophie testiculaire et de gynécomastie ont été également décrits avec dautres agonistes de la GnRH.
Le traitement doit être immédiatement interrompu si le patient présente un quelconque signe ou symptôme indiquant une anaphylaxie/une réaction anaphylactique (dyspnée, asthme, rhinite, dème angioneurotique ou de la glotte, hypotension, urticaire, éruption cutanée, prurit ou pneumonie interstitielle). Les patients doivent être informés avant linstauration du traitement, quen cas dapparition de lun des symptômes mentionnés précédemment, ils doivent arrêter le traitement et consulter leur médecin. Les patients ayant présenté une réaction dhypersensibilité à la leuproréline doivent être étroitement surveillés et ne doivent pas reprendre ZEULIDE 3,75 mg.
Chez les patients traités avec de lacétate de leuproréline, des cas isolés dobstruction urétérale (avec ou sans hématurie) et de compression de la moelle épinière ou de lésions métastatiques vertébrales ont été observés, pouvant entraîner une paralysie avec ou sans complications fatales. Les patients risquant de présenter une obstruction urétérale, une compression de la moelle épinière ou des lésions métastatiques vertébrales doivent être soigneusement examinés et étroitement surveillés durant les premières semaines du traitement. Un traitement prophylactique avec des antiandrogènes doit être envisagé pour ces patients.
Des complications urologiques ou neurologiques peuvent survenir, celles-ci doivent être traitées par des mesures spécifiques appropriées.
Il existe un risque accru dépisode de dépression (pouvant être sévère) chez les patients sous traitement par agonistes de la GnRH, tels que lacétate de leuproréline. Les patients doivent être informés en conséquence et convenablement traités si des symptômes apparaissent.
Une diminution de la densité osseuse a été rapportée dans la littérature médicale chez les hommes ayant subi une orchidectomie ou ayant été traités par un agoniste de la GnRH. Lajout dun antiandrogène au traitement peut réduire la perte osseuse mais augmente le risque dautres effets indésirables tels que des problèmes de coagulation et des dèmes. Lorsquun antiandrogène est utilisé sur une longue période, il convient dapporter une attention particulière aux contre-indications et aux précautions associées à son utilisation prolongée. Les patients à risque ou avec des antécédents dostéoporose doivent être soigneusement examinés et étroitement surveillés pendant le traitement par acétate de leuproréline (voir rubrique 4.8).
Des cas de dysfonctionnement hépatique et de jaunisse accompagnés dun taux élevé denzymes hépatiques ont été rapportés avec lutilisation de lacétate de leuproréline. Par conséquent, une surveillance étroite et les mesures nécessaires doivent être mises en place si nécessaire.
La réponse au traitement par ZEULIDE 3,75 mg doit être surveillée en procédant à des examens cliniques et à lanalyse régulière des taux sériques de testostérone et du PSA.
Certains patients peuvent présenter des changements métaboliques (par ex. intolérance au glucose ou aggravation dun diabète existant), une hypertension, des changements de poids et des troubles cardiovasculaires. Comme attendu avec ce type de médicament, un développement ou une aggravation du diabète peut survenir et par conséquent, les patients diabétiques peuvent nécessiter dune surveillance de la glycémie plus fréquente pendant le traitement par ZEULIDE 3,75 mg. Les patients à haut risque de présenter des maladies métaboliques ou cardiovasculaires doivent être soigneusement évalués avant de commencer le traitement et les patients correctement surveillés pendant le traitement par privation androgénique. Le traitement par acétate de leuproréline entraîne une suppression du système hypophyso-gonadique. Les résultats des tests diagnostiques des fonctions de la gonadostimuline hypophysaire et des gonades effectués pendant et après le traitement par acétate de leuproréline peuvent être altérés.
Une augmentation du temps de prothrombine a été rapportée chez les patients traités avec de lacétate de leuproréline.
Des convulsions ont été rapportées suite à ladministration dacétate de leuproréline. Ces cas ont été observés chez des patients ayant des antécédents de crises de convulsions, dépilepsie, de troubles cérébrovasculaires, danomalies ou de tumeurs du système nerveux central et chez les patients prenant des traitements concomitants ayant été associés aux crises tels que le bupropion et les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS). Des convulsions ont également été rapportées chez des patients en cas dabsence d'une quelconque maladie mentionnée ci-dessus.
Lacétate de leuproréline doit être utilisé avec précaution en cas de maladie cardiovasculaire (y compris linsuffisance cardiaque congestive), de thromboembolie, d'dème, de dépression et dapoplexie hypophysaire.
Lacétate de leuproréline doit être utilisé avec précaution chez les patients présentant des troubles sanguins, une thrombocytopénie ou prenant un traitement anticoagulant.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par flacon, c'est-à-dire quil est essentiellement « sans sodium ».
Une thérapie par privation androgénique peut allonger lintervalle QT
Chez les patients ayant des antécédents ou des facteurs de risque dallongement de lintervalle QT, de même que chez les patients traités en concomitance avec des substances connues pour allonger lintervalle QT (voir rubrique 4.5), les médecins doivent évaluer le rapport bénéfices-risques, y compris la possibilité de torsades de pointe, avant dinstaurer un traitement par ZEULIDE 3,75 mg.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Étant donné quune thérapie par privation androgénique peut allonger lintervalle QT, lutilisation concomitante de ZEULIDE 3,75 mg avec des médicaments connus pour allonger lintervalle QT ou des produits capables dinduire une torsade de pointe, tels que les antiarythmiques de classe IA (ex : quinidine, disopyramide) ou de classe III (ex : amiodarone, sotalol, dofétilide, ibutilide), méthadone, moxifloxacine, antipsychotiques, etc. devra être soigneusement évaluée (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
ZEULIDE 3,75 mg nest pas indiqué chez la femme.
L'injection dacétate de leuproréline peut avoir des effets néfastes sur le ftus lorsquelle est administrée pendant la grossesse.
Par conséquent, un avortement spontané peut se produire si le médicament est administré pendant la grossesse.
ZEULIDE 3,75 mg ne doit pas être utilisé chez les femmes allaitantes.
Fertilité
Les études sur l'animal ont mis en évidence une reprotoxicité (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Sauf indication contraire, le profil de sécurité suivant de ZEULIDE 3,75 mg est basé sur les résultats dun essai clinique de phase III dans lequel des patients atteints dun cancer de la prostate ont été traités avec six doses intramusculaires de ZEULIDE 3,75 mg, une fois par mois, et suivis pendant une période de 26 semaines. La plupart des effets indésirables rapportés liés au traitement étaient ceux généralement associés aux traitements de suppression de la testostérone.
Les effets indésirables les plus fréquemment rapportés liés à ZEULIDE 3,75 mg incluent des bouffées de chaleur, une douleur au point d'injection, une irritation au point d'injection, des sueurs nocturnes et des céphalées.
Les effets indésirables suivants, rapportés lors détudes cliniques, ont été classés par système dorganes et par ordre décroissant de fréquence (très fréquents : ≥1/10 ; fréquents : ≥1/100 à <1/10 ; peu fréquents : ≥ 1/1 000 à < 1/100 ; rares : ≥ 1/10 000 à < 1/1000 ; très rares : < 1/10 000).
Tableau 1. Nombre et fréquence des effets indésirables pendant le traitement par ZEULIDE 3,75 mg.
|
Classe des systèmes dorganes |
Fréquence |
Effet indésirable |
|
Troubles du métabolisme et de la nutrition |
Fréquent |
Augmentation de lappétit |
|
Peu fréquent |
Anorexie, hypercholestérolémie, hyperlipidémie |
|
|
Affections psychiatriques |
Peu fréquent |
Troubles du sommeil, insomnie, baisse de la libido, sautes d'humeur et dépression* |
|
Affections du système nerveux |
Fréquent |
Céphalée |
|
Peu fréquent |
Somnolence |
|
|
Affections de l'oreille et du labyrinthe |
Peu fréquent |
Vertiges |
|
Affections vasculaires |
Très fréquent |
Bouffées de chaleur |
|
Affections gastro-intestinales |
Peu fréquent |
Douleur dans le bas-ventre, diarrhées, nausées, vomissements |
|
Affections hépatobiliaires |
Peu fréquent |
Hyperbilirubinémie |
|
Affections de la peau et du tissu sous-cutané |
Fréquent |
Hyperhidrose, sueurs nocturnes, sueurs froides |
|
Peu fréquent |
dème périorbital, urticaire, prurit |
|
|
Affections musculo-squelettiques et systémiques |
Fréquent |
Douleur dorsale |
|
Peu fréquent |
Arthralgie, spasmes musculaires, douleur dans les extrémités |
|
|
Affections du rein et des voies urinaires |
Peu fréquent |
Rétention urinaire, incontinence urinaire, pollakiurie |
|
Affections des organes de reproduction et du sein |
Fréquent |
Dysfonctionnement érectile |
|
Peu fréquent |
Gonflement de la poitrine, poitrine sensible, absence déjaculation |
|
|
Affections cardiovasculaires |
Fréquence indéterminée |
Allongement de lintervalle QT (voir rubriques 4.4 et 4.5). |
|
Troubles généraux et anomalies au point d'administration |
Fréquent |
Fatigue, asthénie, pyrexie, effets indésirables locaux (voir tableau 2) |
|
Peu fréquent |
Faiblesse, sensation de chaud et froid, sensation de nervosité |
|
|
Affections respiratoires, thoraciques et médiastinales |
Fréquence indéterminée |
Pneumopathie interstitielle |
|
Investigations |
Peu fréquent |
Augmentation de laspartate amino transférase, augmentation de lalanine amino transférase, augmentation de la bilirubine, augmentation de la gamma glutamyl transférase |
*Lors dune étude post-commercialisation, les sautes dhumeur et la dépression étaient des réactions fréquentes chez les utilisateurs à long terme du médicament.
En termes de gravité, 98 % de lensemble des effets indésirables liés au traitement étaient légers ou modérés. Quatre-vingt-neuf pour cent (89 %) des bouffées de chaleur rapportées étaient légères et neuf pour cent (9 %) étaient modérées. Deux cas de bouffées de chaleur (0,2 %) rapportées étaient graves.
Un total de 35 effets indésirables locaux au point dinjection ont été signalés chez 29 patients (18,1 %) au cours de létude.
Les effets indésirables locaux suite à ladministration de ZEULIDE 3,75 mg sont ceux généralement rapportés avec d'autres médicaments similaires administrés par injection intramusculaire. Les douleurs, irritations, gênes, contusions et érythèmes au point d'injection sont les effets indésirables qui ont été rapportés le plus fréquemment. Des effets peu fréquents étaient une réaction au point dinjection, un gonflement, une lésion et une hémorragie (Tableau 2).
Tableau 2. Fréquence des patients présentant des effets indésirables locaux pendant le traitement par ZEULIDE 3,75 mg
|
CSO primaire* Terme préférentiel : Troubles généraux et anomalies au point d'administration |
Patients avec des effets indésirables locaux associés |
|
% |
|
|
Fréquent |
|
|
Douleur au point dinjection |
8,1 |
|
Irritation au point dinjection |
4,4 |
|
Gêne au point dinjection |
1,9 |
|
Érythème au point dinjection |
1,3 |
|
Contusions au point dinjection |
1,3 |
|
|
|
|
Peu fréquents |
|
|
Réaction au point dinjection |
0,6 |
|
Gonflement au point dinjection |
0,6 |
|
Lésion au point dinjection |
0,6 |
|
Hémorragie au point dinjection |
0,6 |
*Les sujets peuvent entrer dans plusieurs catégories ; EIL : effets indésirables locaux ; CSO : classe des systèmes dorganes.
En cas dadministrations répétées de ZEULIDE 3.75 mg, des gonflements (0,6 %), douleurs (0,6 %), contusions (0,6 %) et irritations (0,6 %) ont été rapportés comme étant des effets indésirables locaux récurrents. Ces évènements ont tous été rapportés comme non graves et légers. Aucun patient na arrêté le traitement à cause dévènements indésirables locaux.
Dans un essai clinique de phase I (CRO-02-43) mené chez des sujets sains recevant une dose unique de Leuprolide Depot GP-Pharm 7,5 mg, un seul cas dinduration au point dinjection a été rapporté.
Dautres évènements indésirables ayant été rapportés avec le traitement par acétate de leuproréline incluent limpuissance, une diminution de la libido (deux conséquences pharmacologiques de la privation de testostérone), un dème périphérique, une embolie pulmonaire, des palpitations, une myalgie, une faiblesse musculaire, des frissons, une dyspnée, des vertiges périphériques, une éruption cutanée, lamnésie, des troubles de la vision et une sensibilité cutanée. Linfarctus dun adénome hypophysaire existant a rarement été rapporté suite à ladministration des agonistes de la GnRH à action de longue durée et de courte durée. De rares cas de thrombopénie et de leucopénie ont été rapportés. Des variations de la tolérance au glucose ont été rapportées.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Il ny a aucune expérience clinique des effets dun surdosage significatif de ZEULIDE 3,75 mg ou dacétate de leuproréline. Lors dessais cliniques dans lesquels de lacétate de leuproréline était administré quotidiennement en injection sous-cutanée chez des patients atteints dun cancer de la prostate, des doses atteignant 20 mg/jour pendant deux ans nont causé aucun effet indésirable en dehors de ceux observés avec une dose de 1 mg/jour.
Dans des études menées sur des animaux, des doses allant jusquà 500 fois la dose humaine recommandée ont provoqué une dyspnée, une baisse de lactivité et une irritation locale au point dinjection. En cas de surdosage, le patient doit être étroitement surveillé et sera traité de façon symptomatique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Lacétate de leuproréline est inactif lorsquil est administré par voie orale en raison de sa faible perméabilité membranaire et son inactivation presque complète par les enzymes protéolytiques de lintestin.
Lacétate de leuproréline possède de puissantes propriétés dagoniste de la GnRH lorsquil est administré sur une courte durée et de manière intermittente ; cependant, lorsquil est administré sur une longue durée en continu, les analogues de la GnRH, induisent linhibition de la sécrétion de gonadotrophine et la suppression de la stéroïdogenèse testiculaire.
Lorsquil se lie aux récepteurs hypophysaires de la GnRH, lacétate de leuproréline induit dabord une augmentation des concentrations sériques de lhormone lutéinisante (LH) et de lhormone folliculostimulante (FSH), puis entraîne une augmentation significative des taux de testostérone et de dihydrotestostérone. Néanmoins, cinq à huit jours après leur administration, les analogues de la GnRH induisent une désensibilisation du complexe GnRH-récepteur et/ou une régulation négative de l'hypophyse antérieure. Étant donné quil y a moins de récepteurs sur la surface des cellules, la stimulation cellulaire diminue et une quantité inférieure de gonadotrophine est synthétisée et sécrétée. Après plusieurs semaines de traitement par lagoniste de la GnRH, la sécrétion de LH et de FSH finit par être supprimée. Ainsi, les cellules de Leydig des testicules cessent de produire de la testostérone, et la concentration sérique de testostérone diminue jusqu'à atteindre un niveau correspondant au taux de castration (moins de 0,5 ng/mL) environ deux à quatre semaines après le début du traitement.
Lors dune étude clinique ouverte, multicentrique, à dose multiple portant sur ZEULIDE 3,75 mg, 160 patients atteints dun cancer de la prostate et nayant pas dantécédents de traitement systémique ou dhormonothérapie pour traiter le cancer de la prostate, de chirurgie de la prostate ou dorchidectomie, ont été recrutés. Les objectifs étaient de déterminer lefficacité et la sécurité de ZEULIDE 3,75 mg lorsquil est administré à des patients atteints dun cancer de la prostate qui pourraient bénéficier d'un traitement par privation androgénique. ZEULIDE 3,75 mg a été administré par voie intramusculaire à raison dune dose par mois pendant 6 mois.
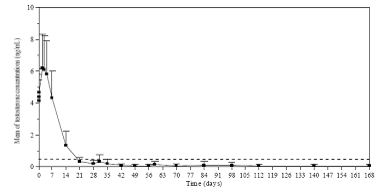
Les taux de testostérone étaient surveillés à plusieurs intervalles pendant 168 jours. Comme attendu, après la première injection les taux moyens de testostérone ont rapidement augmenté par rapport aux taux de départ (4,119 ±1,341 ng/mL), et ont atteint des pics (Cmax) de 6,598 ±2,249 ng/mL dès le troisième jour. Après cette augmentation, les taux de testostérone ont chuté, et le 21e jour, 78,7 % des patients évaluables ont atteint la castration médicale (définie par un taux de testostérone inférieur à 0,5 ng/mL). Le 28e jour, 96,8 % des patients ont atteint des taux correspondant à la castration et 73,1 % ont atteint des taux ≤ 0,2 ng/mL (Figure 1).
Figure 1. Taux plasmatiques moyens de testostérone (±écart type) pendant le traitement administré à raison dune injection IM par mois de ZEULIDE 3,75 mg 3,75 mg pendant six mois
Les critères defficacité secondaires incluaient la détermination des concentrations sériques de LH, FSH et de PSA. Le 14e jour et 4 jours après la première injection de ZEULIDE 3,75 mg, les concentrations sériques moyennes de LH et de FSH avaient atteint un niveau inférieur à celui des concentrations basales. Les concentrations étaient restées largement en dessous des valeurs de départ du 28 e jour jusquà la fin de létude. Pendant le traitement, les taux sériques moyens de PSA ont graduellement diminué (premier mois) puis sont restés constamment en dessous du taux de basal jusqu'à la fin de l'étude. Cependant, une variation importante entre les individus en termes de concentrations de PSA a été observée tout au long de létude.
La fréquence des réponses « acute-on-chronic » a été de 10,5 % et la fréquence de léchappement testostérone a été de 11,8 %. Aucun événement indésirable lié au médicament indiquant une augmentation du taux de testostérone (rétention urinaire, compression de la moelle épinière ou exacerbation de la douleur osseuse) na été rapportée chez les patients présentant un échappement de testostérone.
5.2. Propriétés pharmacocinétiques
Absorption
Après trois injections de ZEULIDE 3,75 mg, à raison dune injection par mois, sur un échantillon de patients atteints de cancer de la prostate (N=12), le pic de concentration plasmatique dacétate de leuproréline était le même pour les trois cycles. Après la première administration (0 au 28e jour), la Cmax était de 13 145,6 ±3 070,6 pg/mL. Le délai médian pour atteindre la Cmax (Tmax) était de 0,04 jours, correspondant à 0,96 h (plage de 0,96 4,08 h).
Distribution
Aucune étude de distribution du médicament na été réalisée avec ZEULIDE 3,75 mg. Cependant, chez des volontaires sains de sexe masculin, le volume moyen de distribution de lacétate de leuproréline à l'état d'équilibre après une administration intraveineuse en bolus de 1,0 mg était de
27 L. In vitro, la liaison aux protéines plasmatiques humaines varie entre 43 % et 49 %.
Biotransformation
Aucune étude du métabolisme du médicament na été réalisée avec ZEULIDE 3,75 mg. Cependant, chez des volontaires de sexe masculin sains, l'administration intraveineuse en bolus de 1 mg dacétate de leuproréline a entraîné une clairance systémique moyenne de 7,6 L/h, avec une demi-vie délimination terminale denviron 3 heures selon un modèle à deux compartiments.
La leuproréline devrait être métabolisée en peptides inactifs plus petits pouvant être excrétés ou catabolisés ultérieurement.
Élimination
Aucune étude dexcrétion du médicament na été réalisée avec ZEULIDE 3,75 mg. Cependant, suite à ladministration dacétate de leuproréline chez 3 patients, moins de 5 % de la dose a été récupérée sous forme de substance mère et de métabolite M-I dans lurine.
Populations particulières
Insuffisance rénale/hépatique
La pharmacocinétique du médicament chez les patients atteints dinsuffisance hépatique et rénale na pas été déterminée.
5.3. Données de sécurité préclinique
Comme attendu aux vues des propriétés pharmacologiques connues, les études non cliniques ont montré des effets réversibles sur le système de reproduction. Dans les études de toxicité pour la reproduction, l'acétate de leuproréline na présenté aucune tératogénicité. Cependant, une embryotoxicité/létalité a été observée chez le lapin.
Des études de cancérogénicité réalisées chez le rat avec lacétate de leuproréline administré par voie sous-cutanée (0,6 à 4 mg/kg/jour), ont montré une augmentation dose-dépendante des adénomes hypophysaires. De plus, une augmentation significative mais non dose-dépendante des adénomes langeaisiens du pancréas chez la femelle et des adénomes des cellules interstitielles testiculaires chez le mâle a été observée, en regardant lincidence la plus élevée dans le groupe recevant de faibles doses. Ladministration dacétate de leuproréline a entraîné linhibition de la croissance de certaines tumeurs hormonodépendantes (tumeurs prostatiques chez des rats Noble et Dunning et tumeurs mammaires induites par le DMBA chez des rats femelles). Aucun effet semblable na été observé dans les études de cancérogénicité réalisées chez la souris. Aucune étude de cancérogénicité na été réalisée avec ZEULIDE 3,75 mg.
L'acétate de leuproréline ne sest pas révélé mutagène lors de tests réalisés in vitro et in vivo. Aucune étude de mutagénicité na été réalisée avec ZEULIDE 3,75 mg.
Excipients du lyophilisat (flacon) :
Polysorbate 80
Mannitol (E421)
Carmellose sodique (E466)
Citrate de triéthyle
Polymère d'acide D,L-lactique et d'acide glycolique (PLGA)
Excipients du solvant (seringue préremplie) :
Mannitol (E421)
Hydroxyde de sodium (pour ajustement du pH)
Acide chlorhydrique (pour ajustement du pH)
Eau pour préparations injectables
Aucun solvant autre que le solvant stérile fourni pour ZEULIDE 3,75 mg ne peut être utilisé pour la reconstitution de la poudre de ZEULIDE 3,75 mg.
Une fois reconstitué avec le solvant, la suspension doit être administrée immédiatement.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C. Ne pas congeler.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
o Un (1) flacon en verre de type I contenant 3,75 mg d'acétate de leuproréline sous forme de poudre lyophilisée, scellé à l'aide d'une capsule en élastomère et d'un bouchon en aluminium muni d'un capuchon amovible en plastique
o Une (1) seringue préremplie en verre de type I contenant 2 mL de solvant, munie dun capuchon en élastomère.
o Un (1) adaptateur en polycarbonate / PEHD et une (1) aiguille stérile de calibre 20 gauge.
6.6. Précautions particulières délimination et de manipulation
Mode d'administration
Le flacon de poudre de microsphères de ZEULIDE 3,75 mg doit être reconstitué immédiatement avant ladministration du médicament par injection intramusculaire. Assurez-vous d'employer une technique aseptique.
La solution reconstituée est une suspension daspect laiteux, de couleur blanche.
Aucun autre solvant ne peut être utilisé pour la reconstitution de ZEULIDE 3,75 mg.
Reconstituer ZEULIDE 3,75 mg conformément aux instructions suivantes :
|
1 |
|
2 |
|
3 |
|
|
|
|
|
|
|
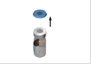
Retirez le capuchon bleu du flacon. |
|
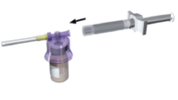
Fixez le système d'adaptation (violet) sur le flacon jusqu'à ce qu'un « clic »se fasse entendre. |
|
Fixez la gâchette blanche sur la seringue contenant le solvant. Retirez le capuchon en élastomère de la seringue et placez cette dernière sur le système d'adaptation. |
|
4 |
|
5 |
|
6 |
|
|
|
|
|
|
|
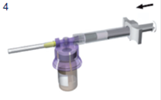
Tout en maintenant la seringue et le flacon solidement connectés, en position verticale, poussez lentement le piston afin de transférer tout le solvant dans le flacon. |
|
La seringue toujours fixée sur le flacon, secouez doucement ce dernier pendant une minute environ, jusqu'à obtention d'une suspension blanche laiteuse homogène. |
|
Retournez l'ensemble et tirez délicatement sur le piston afin d'aspirer le médicament remis en suspension du flacon dans la seringue. |
|
7 |
|
8 |
|
|
|
|
|
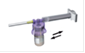
Détachez la seringue et l'aiguille du système d'adaptation en faisant pivoter la partie supérieure de l'adaptateur dans le sens inverse des aiguilles d'une montre. Le médicament est prêt à l'emploi. |
|
Nettoyez le point d'injection à l'alcool et laissez la peau sécher. Injectez la suspension par voie intramusculaire dans le quart supérieur externe du muscle fessier. |
Une partie du produit peut former des agrégats ou sagglomérer sur les parois du flacon. Ce phénomène est normal. Pendant la fabrication du médicament, le flacon est rempli avec un excès de produit afin dassurer que la dose finale de 3,75 mg dacétate de leuproréline soit administrée.
Ce médicament est destiné à une seule injection. Toute solution restante non utilisée doit être éliminée.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
POLIGONO INDUSTRIAL ELS VINYETS ELS FOGARS. SECTOR 2
CARRETERA COMARCAL 244, KM 22
SANT QUINTI DE MEDIONA
08777 BARCELONA
ESPAGNE
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 301 662 4 6 : poudre en flacon (verre) + 2 mL de solvant en seringue préremplie (verre), boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I