
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 15/04/2019
SPORANOX 10 mg/ml, solution buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour 100 ml de solution.
1 dose de 10 ml de solution correspond à 100 mg d'itraconazole.
Excipients à effet notoire : sorbitol
Pour la liste complète des excipients, voir rubrique 6.1.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Traitement des candidoses orales et/ou sophagiennes: 200 mg (20 ml) par jour de préférence en 2 prises, ou éventuellement en 1 prise pendant 1 semaine. En l'absence de réponse après 1 semaine, le traitement doit être poursuivi pendant une semaine supplémentaire.
Traitement des candidoses orales et/ou sophagiennes résistantes au fluconazole: 100 à 200 mg (10 à 20 ml) 2 fois par jour pendant 2 semaines. En l'absence de réponse après deux semaines de traitement, le traitement doit être poursuivi pendant 2 semaines supplémentaires. En l'absence de signes d'amélioration clinique, la dose de 400 mg par jour ne doit pas être utilisée pendant une durée supérieure à 14 jours.
Mode dadministration
Afin d'assurer une absorption optimale, la prise de ce médicament doit s'effectuer en dehors des repas.
La solution doit être laissée en contact avec la bouche pendant quelques instants (environ 20 secondes), puis avalée. Ne pas rincer après avoir avalé (il est recommandé aux patients de s'abstenir de toute prise alimentaire pendant au moins 1 heure après l'administration).
Populations particulières
Utilisation dans la population pédiatrique
Les données cliniques disponibles chez les enfants âgés de moins de 18 ans étant limitées, ce médicament ne sera utilisé chez ces patients que si le bénéfice attendu lemporte sur les risques potentiels (voir rubrique 5.2).
Utilisation chez le sujet âgé
Les données cliniques sur lutilisation de SPORANOX solution buvable chez les patients âgés étant limitées, ce médicament ne sera utilisé chez ces patients que si le bénéfice attendu lemporte sur les risques potentiels.
Utilisation chez l'insuffisant hépatique
Les données disponibles sur lutilisation de litraconazole par voie orale chez les patients insuffisants hépatiques sont limitées. La prudence est recommandée lors de ladministration de ce médicament à cette population de patients (voir rubrique 4.4 et 5.2).
Utilisation chez l'insuffisant réna
Lhémodialyse ou la dialyse rétropéritonéale résulte en une extraction minimale de litraconazole.
Les données disponibles sur lutilisation de litraconazole par voie orale chez les patients insuffisants rénaux sont limitées. Lexposition à litraconazole peut être plus faible chez certains patients atteints dune insuffisance rénale. La prudence est recommandée lors de ladministration de ce médicament à cette population de patients et un ajustement de la dose peut être envisagé (voir rubrique 4.4 et 5.2).
· SPORANOX solution buvable ne doit pas être administré aux patients ayant une dysfonction ventriculaire démontrée tels quune insuffisance cardiaque congestive ou des antécédents dinsuffisance cardiaque congestive sauf en cas dinfections sévères ou mettant en jeu le pronostic vital. (voir rubrique 4.4.)
· La co-administration dun grand nombre de substrats du CYP3A4 est contre-indiquée avec SPORANOX solution buvable. Une augmentation de la concentration plasmatique de ces médicaments, provoquée par la co-administration avec litraconazole, peut augmenter ou prolonger les effets thérapeutiques ou indésirables à un tel niveau que des situations potentiellement graves peuvent survenir. Par exemple laugmentation de la concentration plasmatique de certains de ces médicaments peut entrainer un allongement de lintervalle QT et des tachycardies ventriculaires incluant la survenue de torsades de pointes, une arythmie potentiellement létale. (voir rubrique 4.5)
· SPORANOX solution buvable ne doit pas être administré en association avec : les alcaloïdes de lergot de seigle vasoconstricteurs (tels que dihydroergotamine, ergotamine, méthylergométrine, methysergide), lalfuzosine, laliskiren, latorvastatine, lavanafil, la colchicine (chez les sujets avec insuffisance rénale sévère ou insuffisance hépatique sévère), le dabigatran, la dapoxétine, la dompéridone, la dronédarone, léplérénone, la fésotérodine (chez les sujets ayant une atteinte de la fonction rénale modérée à sévère ou un trouble de la fonction hépatique modéré à sévère), livabradine, le lomitapide, la lurasidone, le millepertuis, la mizolastine, lassociation ombitasvir + paritaprévir, le pimozide, la quétiapine, la quinidine, la ranolazine, le sildénafil (dans lindication du traitement de lhypertension artérielle pulmonaire), la simvastatine, la solifénacine (chez les sujets ayant une insuffisance rénale modérée à sévère ou une insuffisance hépatique modérée à sévère), la télithromycine (chez les sujets ayant une insuffisance rénale sévère ou une insuffisance hépatique sévère), le ticagrélor, le vardénafil (chez lhomme de plus de 75 ans) (voir rubrique 4.5).
· SPORANOX solution buvable ne doit pas être utilisé pendant la grossesse pour des pathologies ne mettant pas en jeu le pronostic vital (voir rubrique 4.6).
Les femmes en âge de procréer, et traitées par SPORANOX doivent utiliser un moyen de contraception efficace. Une contraception efficace doit être poursuivie jusquau début des règles suivant larrêt du traitement par SPORANOX.
4.4. Mises en garde spéciales et précautions d'emploi
De très rares cas de toxicité hépatique grave, incluant quelques cas dinsuffisance hépatique aiguë dévolution fatale ont été rapportés chez des patients traités par SPORANOX. La plupart de ces cas sont survenus chez des patients ayant une maladie hépatique pré-existante, traités pour des mycoses systémiques, ayant des pathologies concomitantes significatives et/ou traités par dautres médicaments hépatotoxiques. Quelques patients ne présentaient pas de facteurs de risque patents de maladie hépatique. Quelques cas ont été observés au cours du 1er mois de traitement, et pour certains au cours de la 1ère semaine de traitement. Il est recommandé de surveiller la fonction hépatique chez les patients traités par SPORANOX. Les patients doivent être informés quils doivent avertir très rapidement leur médecin en cas dapparition de signes et de symptômes suggérant une atteinte hépatique en particulier anorexie, nausées, vomissements, asthénie, douleurs abdominales ou urines foncées. En cas de survenue de lun de ces symptômes, le traitement doit être immédiatement interrompu et des examens de la fonction hépatique doivent être réalisés.
Chez les patients ayant une élévation des enzymes hépatiques ou présentant une maladie hépatique active, ou chez qui d'autres médicaments ont déjà entraîné une toxicité hépatique, le traitement ne doit pas être débuté à moins que le patient se trouve dans une situation grave ou mettant en jeu le pronostic vital pour laquelle le bénéfice attendu de SPORANOX lemporte sur le risque datteinte hépatique. Si le traitement est débuté, la fonction hépatique doit être surveillée.
Effets cardiaques
Une étude réalisée chez le volontaire sain avec SPORANOX IV a montré une diminution transitoire asymptomatique de la fraction d'éjection ventriculaire gauche ; se résolvant avant la perfusion suivante. La signification clinique de cette observation, pour les formes orales, n'est pas connue.
L'itraconazole a montré un effet inotrope négatif et SPORANOX a été associé à des cas d'insuffisance cardiaque congestive après administration par voie orale.
Dans les notifications spontanées, les insuffisances cardiaques ont été plus fréquemment rapportées lorsque la dose journalière totale ditraconazole était de 400 mg par rapport à une dose journalière totale plus faible, ce qui suggère que le risque dinsuffisance cardiaque peut être dose-dépendant.
SPORANOX ne doit être utilisé chez des patients présentant une insuffisance cardiaque congestive ou des antécédents d'insuffisance cardiaque congestive que si le bénéfice est nettement supérieur aux risques. L'évaluation individuelle du rapport bénéfice/risque doit prendre en compte des facteurs tels que la sévérité de l'indication, la posologie (i.e. la dose totale journalière) et les facteurs de risques individuels d'insuffisance cardiaque congestive. Ces facteurs de risque comprennent des maladies cardiaques telles que les maladies ischémiques et valvulaires, des maladies pulmonaires significatives telles que la bronchopneumopathie chronique obstructive, l'insuffisance rénale et d'autres troubles démateux. Ces patients doivent être informés des signes et symptômes de l'insuffisance cardiaque congestive, ils doivent être traités avec précautions et doivent faire l'objet d'un suivi des signes et symptômes de l'insuffisance cardiaque congestive au cours du traitement. Si de tels signes ou symptômes apparaissent au cours du traitement, SPORANOX doit être arrêté.
Les inhibiteurs calciques peuvent avoir des effets inotropes négatifs qui peuvent s'ajouter à ceux de l'itraconazole ; l'itraconazole peut inhiber le métabolisme des inhibiteurs calciques. En conséquence, la prudence s'impose en cas de co-administration d'itraconazole et d'inhibiteurs calciques en raison dun risque augmenté dinsuffisance cardiaque congestive (voir rubrique 4.5).
Hypersensibilité croisée
Les informations relatives à une hypersensibilité croisée entre litraconazole et dautres antifongiques azolés sont limitées. La prudence est de rigueur lorsque ce médicament est prescrit à des patients ayant présenté une hypersensibilité à dautres azolés.
Excipients à effet notoire
Ce médicament contient du sorbitol. Son utilisation est déconseillée chez les patients présentant une intolérance au fructose (maladie héréditaire rare).
Allaitement
Lallaitement est déconseillé en cas de traitement par ce médicament.
Interactions médicamenteuses
Ce médicament est déconseillé dans les cas suivants en association avec : lapixaban, la bédaquiline, la buspirone, le busulfan, la colchicine (sauf chez les sujets avec insuffisance rénale sévère ou insuffisance hépatique sévère : association contre-indiquée, voir rubrique 4.5), la darifénacine, lébastine les inhibiteurs des tyrosines kinases suivants : [axitinib, bosutinib, dabrafénib, dasatinib, ibrutinib, nilotinib, sunitinib], lhalofantrine, lhalopéridol, les immunosuppresseurs [ciclosporine, évérolimus, sirolimus, tacrolimus, temsirolimus], les inducteurs enzymatiques suivants : [carbamazépine, dabrafénib, éfavirenz, enzalutamide, esclicarbazépine, fosphénytoïne, névirapine, oxcarbazépine, phénobarbital, phénytoïne, primidone, rifabutine, rifampicine], lirinotécan, la lercanidipine, la luméfantrine, le midazolam per os, loxycodone, le régorafénib, le rivaroxaban, le riociguat, le siméprévir, le tadalafil (dans le traitement de lhypertension artérielle pulmonaire ou des symptômes et signes de lhypertrophie bénigne de la prostate), la tamsulosine, la toltérodine, la trabectédine, le vardénafil (chez lhomme jusquà 75 ans) et les vinca-alcaloïdes cytotoxiques (voir rubrique 4.5).
Insuffisance rénale
Lhémodialyse ou la dialyse rétropéritonéale résulte en une extraction minimale de litraconazole.
Les données disponibles sur lutilisation de litraconazole par voie orale chez les insuffisants rénaux sont limitées. Lexposition à litraconazole peut être plus faible chez certains patients atteints dune insuffisance rénale. La prudence est recommandée lors de ladministration de ce médicament à cette population de patients et un ajustement de la dose peut être envisagé (voir rubriques 4.2 et 5.2).
Insuffisance hépatique
Les données relatives à lutilisation de litraconazole par voie orale chez les patients insuffisants hépatiques sont limitées. La prudence est recommandée lors de ladministration de ce médicament à cette population de patients. Il est préconisé de surveiller étroitement les patients insuffisants hépatiques en cas de prise ditraconazole (voir rubrique 4.2).
Lors de la décision dinitier un traitement avec dautres médicaments métabolisés par le CYP3A4, il est recommandé de prendre en compte la prolongation de la demi-vie délimination de litraconazole observée dans un essai clinique conduit avec litraconazole administré à dose unique sous forme de gélules chez des patients cirrhotiques (voir rubrique 5.2).
Sujets immunodéprimés (par exemple : patients neutropéniques, patients infectés par le VIH+)
Les concentrations à létat stationnaire sont généralement plus faibles chez les immunodéprimés et justifient lutilisation de posologies élevées (400 mg/jour). La surveillance des concentrations plasmatiques peut être utile en début de traitement surtout sil existe des éléments susceptibles de modifier labsorption (prise à jeun, réaction du greffon contre lhôte, diarrhée, mucite), et en cas de suspicion déchec.
Traitement des patients atteints de neutropénie sévère
Ce médicament, dans le traitement des candidoses orales et/ou sophagiennes, na pas été étudié chez les patients atteints de neutropénie sévère. Du fait de ses propriétés pharmacocinétiques (voir rubrique 5.2), ce médicament nest pas recommandé en initiation de traitement chez des patients présentant un risque immédiat de candidose systémique.
Mucoviscidose
Chez les patients atteints de mucoviscidose, une variabilité des concentrations plasmatiques ditraconazole a été observée à létat déquilibre avec la solution buvable ditraconazole administrée à la posologie de 2,5 mg/kg 2 fois par jour. A létat déquilibre, des concentrations supérieures à 250 ng/ml ont été atteintes chez environ 50 % des patients de plus de 16 ans mais chez aucun patient de moins de 16 ans. En labsence de réponse au traitement par SPORANOX, solution buvable une alternative thérapeutique doit être envisagée.
Perte auditive
Des cas de perte auditive transitoire ou permanente ont été rapportés chez des patients traités par de litraconazole. Plusieurs de ces cas comprenaient ladministration concomitante de quinidine, qui est une association contre-indiquée (voir rubriques 4.3 et 4.5). La perte auditive se dissipe généralement à larrêt du traitement, mais elle peut persister chez certains patients.
Neuropathie
En cas dapparition dune neuropathie qui pourrait être imputée à ce médicament, il convient dinterrompre le traitement.
Résistance croisée
En cas de candidoses systémiques, si des souches de Candida sont suspectées résistantes au fluconazole, on ne peut prédire leur sensibilité à l'itraconazole; donc celle-ci devra être testée avant dinitier le traitement par litraconazole.
Interchangeabilité
Il nest pas recommandé dinterchanger SPORANOX gélule et SPORANOX solution buvable car pour une même dose administrée lexposition à ce médicament est supérieure avec la solution buvable quavec les gélules.
Par ailleurs, les effets topiques de lexposition de la muqueuse digestive à litraconazole peuvent être différents entre les 2 formulations. Seule la solution buvable est indiquée dans le traitement de la candidose orale/et /ou sophagienne chez les patients infectés par le VIH.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Associations contre-indiquées (voir rubrique 4.3)
+ Alcaloïdes de lergot de seigle vasoconstricteurs (tels que dihydroergotamine, ergotamine, méthylergométrine et méthysergide) :
Risque de vasoconstriction coronaire ou des extrémités (ergotisme), ou de poussées hypertensives.
+ Alfuzosine
Risque daugmentation des concentrations plasmatiques de lalfuzosine et de ses effets indésirables.
+ Aliskiren
Augmentation, de près de 6 fois des concentrations plasmatiques daliskiren et majoration du risque de ses effets indésirables.
+ Atorvastatine
Risque majoré deffets indésirables (concentration-dépendants) à type de rhabdomyolyse (diminution du métabolisme hépatique de latorvastatine).
+ Avanafil
Augmentation très importante des concentrations plasmatiques de lavanafil avec risque dhypotension.
+ Colchicine chez les sujets avec insuffisance rénale sévère ou insuffisance hépatique sévère
Augmentation des effets indésirables de la colchicine aux conséquences potentiellement fatales.
+ Dabigatran
Augmentation de plus du double des concentrations plasmatiques de dabigatran, avec majoration du risque de saignement.
+ Dapoxétine
Risque de majoration des effets indésirables notamment à type de vertiges ou de syncopes.
+ Dompéridone
Augmentation des concentrations plasmatiques de dompéridone par diminution de son métabolisme hépatique par litraconazole.
+ Dronédarone
Augmentation importante des concentrations de dronédarone par diminution de son métabolisme.
+ Eplérénone
Risque daugmentation des concentrations plasmatiques de léplérénone par litraconazole et de ses effets indésirables, notamment lhyperkaliémie.
+ Fésotérodine chez les sujets avec insuffisance rénale modérée à sévère ou insuffisance hépatique modérée à sévère
Risque de majoration des effets indésirables, notamment à type de troubles du rythme cardiaque.
+ Ivabradine
Augmentation des concentrations plasmatiques de livabradine et par conséquent de ses effets indésirables (inhibition de son métabolisme hépatique par litraconazole).
+ Lomitapide
Augmentation des concentrations plasmatiques du lomitapide par diminution de son métabolisme hépatique par litraconazole.
+ Lurasidone
Augmentation des concentrations plasmatiques de la lurasidone par diminution de son métabolisme hépatique par litraconazole.
+ Millepertuis
Risque de diminution importante des concentrations plasmatiques ditraconazole, avec risque de perte defficacité, par augmentation de son métabolisme hépatique par le millepertuis.
+ Mizolastine
Risque de troubles du rythme ventriculaire, notamment de torsades de pointes.
+ Ombitasvir + Paritaprévir
Augmentation des concentrations plasmatiques de la bithérapie par diminution de son métabolisme hépatique par litraconazole.
+ Pimozide
Risque majoré de troubles du rythme ventriculaire, notamment de torsades de pointes.
+ Quétiapine
Augmentation importante des concentrations de quétiapine, avec risque de surdosage.
+ Quinidine
Risque majoré de troubles du rythme ventriculaire, notamment de torsades de pointes, ainsi que d'acouphènes et/ou de diminution de l'acuité auditive (cinchonisme), par diminution du métabolisme hépatique de la quinidine.
+ Ranolazine
Augmentation des concentrations de ranolazine par diminution de son métabolisme par litraconazole.
+ Sildénafil (dans le traitement de lhypertension artérielle pulmonaire)
Augmentation très importante des concentrations plasmatiques du sildénafil, avec risque dhypotension.
+ Simvastatine
Risque majoré deffets indésirables (concentration-dépendants) à type de rhabdomyolyse (diminution du métabolisme hépatique de la simvastatine).
+ Solifénacine chez les sujets avec insuffisance rénale modérée à sévère ou insuffisance hépatique modérée à sévère
Risque de majoration des effets indésirables, notamment à type de troubles du rythme cardiaques.
+ Télithromycine chez les sujets avec insuffisance rénale sévère ou insuffisance hépatique sévère
Risque de majoration des effets indésirables, notamment à type de troubles du rythme cardiaque.
+ Ticagrélor
Augmentation des concentrations plasmatiques de ticagrélor par diminution de son métabolisme hépatique par litraconazole.
+ Vardénafil (chez lhomme de plus de 75 ans)
Augmentation très importante des concentrations plasmatiques de vardénafil, avec risque dhypotension sévère.
Associations déconseillées (voir rubrique 4.4)
+ Apixaban
Augmentation des concentrations plasmatiques de lapixaban par litraconazole avec majoration du risque de saignement.
+ Bédaquiline
Augmentation des concentrations plasmatiques de bédaquiline par diminution de son métabolisme hépatique par litraconazole.
Si lassociation est nécessaire, une surveillance ECG plus fréquente et une surveillance des transaminases sont recommandées.
+ Buspirone
Augmentation des concentrations plasmatiques de la buspirone par diminution de son métabolisme hépatique, avec majoration importante de la sédation.
+ Busulfan
Avec le busulfan à fortes doses : doublement des concentrations de busulfan par litraconazole.
+ Colchicine (sauf chez les sujets avec insuffisance rénale sévère ou insuffisance hépatique sévère : voir association contre-indiquée)
Augmentation des effets indésirables de la colchicine aux conséquences potentiellement fatales.
+ Darifénacine
Augmentation des concentrations de darifénacine avec risque de majoration de ses effets indésirables.
Surveillance clinique et adaptation éventuelle de la posologie de darifénacine.
+ Ebastine
Risque majoré de troubles du rythme ventriculaire chez les sujets prédisposés (syndrome du QT long congénital).
+ Inhibiteurs des tyrosines kinases : axitinib, bosutinib, dabrafénib, dasatinib, ibrutinib, nilotinib, sunitinib
Pour les autres inhibiteurs de tyrosine kinase, voir en associations faisant lobjet dune précaution demploi
Risque de majoration des effets indésirables de linhibiteur de tyrosine kinase par diminution de son métabolisme.
Avec librutinib, si lassociation ne peut être évitée, adaptation de la posologie dibrutinib ou interruption temporaire (7 jours).
+ Halofantrine
Risque majoré de troubles du rythme ventriculaire, notamment de torsades de pointes. Si cela est possible, interrompre l'azolé antifongique. Si l'association ne peut être évitée, contrôle préalable du QT et surveillance ECG monitorée.
+ Halopéridol
Risque de troubles du rythme ventriculaire notamment de torsade de pointes, par diminution du métabolisme de lhalopéridol par litraconazole.
+ Immunosuppresseurs (ciclosporine, évérolimus, sirolimus, tacrolimus, temsirolimus)
Augmentation très importante des concentrations sanguines de limmunosuppresseur par inhibition de son métabolisme hépatique. En cas dassociation, contrôle strict de la fonction rénale, dosage des concentrations sanguines de limmunosuppresseur et adaptation éventuelle de la posologie
+ Inducteurs enzymatiques (carbamazépine, dabrafénib, éfavirenz, enzalutamide, esclicarbazépine, fosphénytoïne, névirapine, oxcarbazépine, phénobarbital, phénytoïne, primidone, rifabutine, rifampicine)
Diminution des concentrations plasmatiques ditraconazole, avec risque de perte defficacité, par augmentation de son métabolisme hépatique par linducteur.
+ Irinotécan
Risque de majoration des effets indésirables de lirinotécan par augmentation des concentrations plasmatiques de son métabolite actif.
+ Lercanidipine
Risque majoré d'effets indésirables, notamment ddèmes, par diminution du métabolisme hépatique de la dihydropyridine.
+ Luméfantrine
Risque majoré de troubles du rythme ventriculaire, notamment de torsades de pointes. Si cela est possible, interrompre le torsadogène associé. Si lassociation ne peut être évitée, contrôle préalable du QT et surveillance ECG monitorée.
+ Midazolam per os
Augmentation des concentrations plasmatiques de midazolam par diminution de son métabolisme hépatique avec majoration de la sédation.
+ Oxycodone
Augmentation des concentrations plasmatiques de loxycodone. Surveillance clinique et adaptation éventuelle de la posologie doxycodone pendant la durée du traitement par litraconazole.
+ Régorafénib
Augmentation des concentrations plasmatiques de régorafénib par diminution de son métabolisme hépatique par litraconazole.
+ Riociguat
Augmentation des concentrations plasmatiques de riociguat par diminution de son métabolisme hépatique par litraconazole.
+ Rivaroxaban
Augmentation des concentrations plasmatiques de rivaroxaban, avec majoration du risque de saignement.
+ Siméprévir
Risque daugmentation des concentrations plasmatiques de siméprévir par diminution de son métabolisme hépatique par litraconazole.
+ Tadalafil (dans le traitement de lhypertension artérielle pulmonaire ou des symptômes et signes de lhypertrophie bénigne de la prostate)
Augmentation des concentrations plasmatiques du tadalafil, avec risque dhypotension.
+ Tamsulosine
Risque de majoration des effets indésirables de la tamsulosine, par inhibition de son métabolisme hépatique.
+ Toltérodine
Risque de majoration des effets indésirables.
+ Trabectédine
Risque daugmentation des concentrations plasmatiques de la trabectédine par litraconazole. Si lassociation est nécessaire, surveillance clinique et adaptation éventuelle de la posologie de la trabectédine pendant la durée du traitement par litraconazole.
+ Vardénafil (chez lhomme jusquà 75 ans)
Augmentation très importante des concentrations plasmatiques de vardénafil, avec risque dhypotension sévère.
+ Vinca-alcaloïdes cytotoxiques
Majoration de la neurotoxicité de lantimitotique, par diminution de son métabolisme hépatique par litraconazole.
Associations faisant l'objet de précautions demploi
+ Alfentanil, fentanyl
Augmentation de leffet dépresseur respiratoire de lanalgésique opiacé par diminution de son métabolisme hépatique. Surveillance clinique et adaptation de la posologie de lanalgésique opiacé en cas de traitement par litraconazole.
+ Antagonistes des canaux calciques (diltiazem, vérapamil, classe des dihydropyridines) sauf lercanidipine (voir en associations déconseillées)
Majoration des effets indésirables de lantagoniste des canaux calciques, le plus souvent à type dhypotension notamment chez le sujet âgé. Surveillance clinique et adaptation posologique pendant le traitement par litraconazole et après son arrêt.
De plus avec le vérapamil, bradycardie et/ou troubles de la conduction auriculo-ventriculaire, par diminution du métabolisme hépatique du vérapamil par litraconazole. Surveillance clinique et ECG. Sil y a lieu, adaptation de la posologie du vérapamil pendant le traitement par litraconazole, et après son arrêt, le cas échéant.
+ Antivitamines K
Augmentation de l'effet de l'antivitamine K et du risque hémorragique. Contrôle plus fréquent de l'INR. Adaptation éventuelle de la posologie de l'antivitamine K pendant le traitement par l'itraconazole et 8 jours après son arrêt.
+ Aripiprazole
Risque daugmentation des concentrations plasmatiques daripiprazole par litraconazole. Surveillance clinique et éventuellement adaptation de la posologie daripiprazole pendant et après larrêt du traitement par litraconazole.
+ Bortézomib
Risque de majoration des effets indésirables, notamment neurologiques du bortézomib par diminution de son métabolisme. Surveillance clinique et adaptation éventuelle de la posologie du bortézomib pendant la durée du traitement par litraconazole.
+ Bosentan
Risque majoré des effets indésirables du bosentan, notamment datteintes hépatiques, par diminution de son métabolisme par litraconazole. Surveillance clinique et biologique pendant lassociation.
+ Buprénorphine
Augmentation des concentrations de buprénorphine par diminution de son métabolisme hépatique, avec risque de majoration de ses effets indésirables. Surveillance clinique et adaptation de la posologie de la buprénorphine pendant le traitement par litraconazole et, le cas échéant, après son arrêt.
+ Cabazitaxel
Risque de majoration des effets indésirables dose-dépendants du cabazitaxel par inhibition de son métabolisme par litraconazole Surveillance clinique et adaptation éventuelle de la posologie du cabazitaxel pendant le traitement par litraconazole.
+ Daclatasvir
Augmentation des concentrations de daclatasvir par litraconazole. La dose de daclatasvir doit être diminuée à 30 mg 1 fois par jour en cas de co-administration avec litraconazole.
+ Digoxine
Augmentation de la digoxinémie avec nausées, vomissements, troubles du rythme. Surveillance clinique et, s'il y a lieu, de l'ECG et de la digoxinémie avec adaptation de la posologie de la digoxine pendant le traitement par litraconazole et après son arrêt.
+ Disopyramide
Risque daugmentation des concentrations du disopyramide et de ses effets indésirables par litraconazole. Surveillance clinique et éventuellement adaptation de la posologie du disopyramide.
+ Docétaxel
Risque de majoration des effets indésirables dose-dépendants du docétaxel par inhibition de son métabolisme par litraconazole. Surveillance clinique et adaptation éventuelle de la posologie du docétaxel pendant le traitement par litraconazole.
+ Fésotérodine (sauf chez les sujets avec insuffisance rénale modérée à sévère ou insuffisance hépatique modérée à sévère : voir association contre-indiquée)
Risque de majoration des effets indésirables, notamment à type de troubles du rythme cardiaque. Limiter la dose maximale de fésotérodine à 4 mg par jour lors de la co-administration avec litraconazole.
+ Hydroquinidine
Risque d'acouphènes et/ou de diminution de l'acuité auditive : cinchonisme lié à une diminution du métabolisme hépatique de l'antiarythmique par l'itraconazole. Surveillance des concentrations plasmatiques de l'antiarythmique et diminution éventuelle de sa posologie si nécessaire.
+ Inhibiteurs de protéases boostés par ritonavir ou cobicistat
Risque daugmentation des concentrations plasmatiques de litraconazole. Surveillance clinique. Ladministration de doses élevées ditraconazole nest pas recommandée (>200 mg par jour).
+ Inhibiteurs des tyrosines kinases (sauf bosutinib, ibrutinib, axitinib, dabrafénib, dasatinib, nilotinib, sunitinib : voir en association déconseillée) : cabozantinib, crizotonib, erlotinib, géfitinib, imatinib, lapatinib, pazopanib, ponatinib, ruxolitinib, sorafénib.
Risque de majoration des effets indésirables de linhibiteur de tyrosine kinase par diminution de son métabolisme. Surveillance clinique.
+ Ivacaftor
Augmentation importante des concentrations divacaftor avec risque de majoration des effets indésirables. Réduire la dose du quart, soit 150 mg 1 jour sur 2.
+ Maraviroc
Augmentation des concentrations de maraviroc par litraconazole. La dose de maraviroc doit être diminuée à 150 mg deux fois par jour en cas de co-administration avec litraconazole.
+ Midazolam IV
Augmentation des concentrations plasmatiques de midazolam par diminution de son métabolisme hépatique, avec majoration de la sédation. Surveillance clinique et réduction de la posologie de midazolam en cas de traitement par litraconazole.
+ Quinine
Risque de majoration des effets indésirables de la quinine, notamment troubles du rythme ventriculaire et troubles neurosensoriels (cinchonisme). Surveillance clinique et ECG. Adaptation éventuelle de la posologie de la quinine pendant le traitement par litraconazole et après son arrêt.
+ Sildénafil (dans le traitement de la dysfonction érectile)
Augmentation des concentrations plasmatiques du sildénafil, avec risque dhypotension.
Débuter le traitement par sildénafil à la dose minimale en cas dassociation avec litraconazole.
+ Solifénacine (sauf chez les sujets avec insuffisance rénale modérée à sévère ou insuffisance hépatique modérée à sévère : voir association contre-indiquée)
Risque de majoration des effets indésirables, notamment à type de troubles du rythme cardiaques. Limiter la dose maximale de solifénacine à 5 mg par jour lors de la co-administration avec litraconazole.
+ Sufentanil
Augmentation de leffet dépresseur respiratoire de lanalgésique opiacé par diminution de son métabolisme hépatique. Surveillance clinique et adaptation de la posologie de lanalgésique opiacé en cas de traitement par litraconazole.
+ Tadalafil (dans le traitement de la dysfonction érectile)
Augmentation des concentrations plasmatiques du tadalafil, avec risque dhypotension. Débuter le traitement par tadalafil à la dose minimale en cas dassociation avec litraconazole.
+ Topiques gastro-intestinaux et adsorbants
Risque de diminution de labsorption de litraconazole. Prendre les topiques ou les adsorbants à distance de litraconazole (plus de 2 heures si possible).
Associations à prendre en compte
+ Alprazolam
Possible augmentation de leffet sédatif de lalprazolam.
+ Aprépitant
Augmentation des concentrations daprépitant par diminution de son métabolisme hépatique par litraconazole.
+ Corticoïdes, notamment inhalés (budésonide, fluticasone, mométasone, ciclésonide, dexaméthasone, méthyprednisolone)
En cas dutilisation prolongée par voie orale ou inhalée, augmentation des concentrations plasmatiques du corticoïde par diminution de son métabolisme hépatique par l'itraconazole, avec risque d'apparition d'un syndrome cushingoïde voire dune insuffisance surrénalienne.
+ Idélalisib
Augmentation des concentrations plasmatiques didélalisib par diminution de son métabolisme hépatique par litraconazole.
+ Oxybutynine
Risque de majoration des effets indésirables de loxybutynine.
+ Salmétérol
Augmentation importante des concentrations de salmétérol par diminution de son métabolisme hépatique par litraconazole.
+ Venlafaxine
Augmentation des concentrations de venlafaxine avec risque de surdosage.
+ Zolpidem
Légère augmentation de leffet sédatif du zolpidem.
+ Zopiclone
Légère augmentation de leffet sédatif de la zopiclone.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études effectuées chez lanimal ont mis en évidence un effet tératogène (voir rubrique 5.3).
Il a été montré que litraconazole traverse le placenta dans un modèle chez le rat.
Des cas d'anomalies congénitales ont été rapportés au cours de la commercialisation de SPORANOX, sans quaucun lien de cause à effet avec la prise de SPORANOX n'ait été établi. Ces cas incluaient notamment des malformations du squelette et des altérations chromosomiques.
Les données cliniques sur l'utilisation de SPORANOX pendant la grossesse sont globalement limitées. Par conséquent, SPORANOX solution buvable ne doit pas être utilisé pendant la grossesse, sauf dans les pathologies mettant en jeu le pronostic vital, où le bénéfice potentiel pour la mère l'emporte sur le préjudice potentiel pour le ftus (voir rubrique 4.3).
Femmes en âge de procréer
Les femmes en âge de procréer, et traitées par SPORANOX, solution buvable doivent utiliser un moyen de contraception efficace. Une contraception efficace doit être poursuivie jusquau début des règles suivant larrêt du traitement par SPORANOX.
Allaitement
En raison de lexcrétion de litraconazole dans le lait maternel et compte-tenu de son profil deffets secondaires, lallaitement est déconseillé en cas de traitement par ce médicament.
Fertilité
Les données animales chez le rat nont pas mis en évidence un effet de litraconazole sur la fertilité mâle ou femelle.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de tolérance
Les effets indésirables les plus fréquemment rapportés au cours du traitement par SPORANOX solution buvable dans les essais cliniques et/ou à partir des notifications spontanées ont été sensation de vertige, céphalées, dysgueusie, dyspnée, toux, troubles gastro-intestinaux (diarrhée, nausée, vomissement, douleur abdominale, dyspepsie), rash et pyrexie. Les effets indésirables les plus sévères ont été les réactions allergiques sévères, linsuffisance cardiaque/linsuffisance cardiaque congestive/dème pulmonaire, les pancréatites, lhépatotoxicité sévère (incluant quelques cas dinsuffisance hépatique aiguë dissue fatale) et réactions cutanées sévères. Se référer à la sous-rubrique « Liste tabulée des effets indésirables » pour les fréquences et pour les autres effets indésirables observés. Se référer à la rubrique 4.4 pour des informations complémentaires sur les autres effets sévères.
Liste tabulée des effets indésirables
Les effets indésirables décrits dans le tableau suivant ont été observés lors des études cliniques réalisées avec SPORANOX solution buvable chez 889 patients dans le traitement des candidoses sophagiennes et oropharyngées, et/ou sont issus des notifications spontanées rapportées après la commercialisation de litraconazole, toute formulation confondue.
Le tableau ci-dessous présente les effets indésirables classés par Système organe et fréquence selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classe de systèmes organes |
Effets indésirables |
||
|
Fréquent (≥ 1/100 à < 1/10) |
Peu fréquent (≥ 1/1 000 à < 1/100) |
Fréquence indéterminée |
|
|
Affections hématologiques et du système lymphatique |
|
Leucopénie, Neutropénie, Thrombopénie |
|
|
Affections du système immunitaire |
|
Hypersensibilité* |
Maladie sérique, dème de Quincke, Réaction anaphylactiques et anaphylactoïdes |
|
Troubles du métabolisme et de la nutrition |
|
Hypokaliémie |
Hypertriglycéridémie, |
|
Affections du système nerveux |
Sensation vertigineuse, Céphalées, Dysgueusie |
Neuropathie périphérique*, Paresthésie, Hypoesthésie |
Tremblements, Vertiges** |
|
Affections oculaires |
|
Troubles visuels (incluant diplopie et vision floue) |
|
|
Affections de l'oreille et du labyrinthe |
|
Acouphène |
Perte auditive passagère ou permanente* |
|
Affections cardiaques |
|
Insuffisance cardiaque |
Insuffisance cardiaque congestive* |
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée, Toux |
|
dème pulmonaire, |
|
Affections gastro-intestinales |
Douleurs abdominales, Diarrhées, Vomissements, Nausées, Dyspepsie |
Constipation |
Pancréatite, Flatulence** |
|
Affections hépatobiliaires |
|
Augmentation des enzymes hépatiques (notamment ALAT, ASAT), Insuffisance hépatique*, Hépatite*, Hyperbilirubinémie |
Hépatotoxicité sévère (incluant quelques cas dinsuffisance hépatique aiguë dissue fatale)* |
|
Affections de la peau et du tissu sous-cutané |
Rash |
Urticaire, Prurit |
Nécrolyse épidermique toxique, Syndrome de Stevens-Johnson, Pustulose exanthématique aiguë généralisée, Erythème polymorphe, Dermatite exfoliative, Vascularite leucocytoclasique, Alopécie, Photosensibilité, |
|
Affections musculo-squelettiques et systémiques |
|
Myalgie, Arthralgie |
|
|
Affections du rein et des voies urinaires |
|
|
Pollakiurie, Incontinence urinaire |
|
Affections des organes de reproduction et du sein |
|
Troubles menstruels |
Dysfonction érectile |
|
Troubles généraux et anomalies au site d'administration |
Pyrexie |
dème |
|
|
Investigations |
|
|
Augmentation de la créatine phosphokinase sanguine |
* Voir rubrique 4.4
** Effet(s) indésirables associés à litraconazole dans les essais cliniques avec SPORANOX gélule
Population pédiatrique
Le profil de sécurité demploi de SPORANOX solution buvable a été évalué chez 250 patients pédiatriques âgés de 6 mois à 14 ans ayant participé à cinq essais cliniques en ouvert.
Sur la base des données de sécurité poolées issues de ces essais cliniques, les effets indésirables très fréquemment rapportés chez les patients pédiatriques ayant reçu au moins une dose de SPORANOX solution buvable en prophylaxie des infections fongiques ou pour le traitement dun muguet buccal ou dinfections fongiques systémiques ont été vomissement (36,0%), pyrexie (30,8%), diarrhée (28,4%), inflammation des muqueuses (23,2%), rash (22,8%), douleur abdominale (17,2%), nausée (15,6%), hypertension (14,0%) et toux (11,2%). La nature des effets indésirables chez les patients pédiatriques est similaire à celle observée chez les patients adultes, mais la fréquence est plus élevée chez les patients pédiatriques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.ansm.sante.fr.
De manière générale, les effets indésirables rapportés en cas de surdosage ont été cohérents avec ceux rapportés lors de lutilisation de litraconazole (voir rubrique 4.8).
Traitement
En cas de surdosage, un traitement symptomatique sera mis en place.
Du charbon activé peut être administré, si nécessaire.
L'itraconazole n'est pas éliminé par hémodialyse.
Il n'existe aucun antidote spécifique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antimycosiques à usage systémique, dérivés triazolés
Code ATC : J02A C02
Mécanisme daction
Litraconazole inhibe lenzyme fongique lanosterol 14 alpha-demethylase (CYP51), qui catalyse une étape indispensable de la biosynthèse de lergostérol, un constituant essentiel de la membrane fongique.
Relation pharmacocinétique (PK) / pharmacodynamique (PD)
Pour les agents triazolés antifongiques, incluant litraconazole, le ratio de laire sous la courbe des concentrations en fonction du temps/concentration minimale inhibitrice (ASC/CMI) est le paramètre le plus étroitement associé à lefficacité.
Microbiologie
Litraconazole a démontré être actif in vitro contre les microorganismes suivants :
Candida spp. (dont Candida albicans, C. tropicalis, C. parapsilosis et Candida dubliniensis).
Candida krusei, Candida glabrata et Candida guillermondii sont généralement les espèces de Candida les moins sensibles, avec quelques isolats ayant montré in vitro une résistance sans équivoque à litraconazole
Résistance
Les principaux mécanismes de résistance incluent lacquisition de mutations entrainant la surexpression du gène codant pour lenzyme cible lanosterol 14 alpha-demethylase, des mutations ponctuelles résultant en une substitution dacides aminés de lenzyme cible conduisant à une diminution de laffinité pour la cible et/ou une surexpression du transporteur, responsables dune augmentation de lefflux.
Concentrations critiques
Les concentrations minimales inhibitrices critiques établies par la méthode EUCAST pour litraconazole sont:
Candida albicans: S ≤0.06 mg/L, R >0.06 mg/L
Candida parapsilosis: S ≤0.12 mg/L, R >0.12 mg/L
Candida tropicalis: S ≤0.12 mg/L, R >0.12 mg/L
5.2. Propriétés pharmacocinétiques
Caractéristiques pharmacocinétiques générales
Le pic plasmatique des concentrations ditraconazole est atteint 2,5 heures après administration de la solution buvable.
En raison de sa pharmacocinétique non linéaire, litraconazole saccumule dans le plasma après administrations répétées. Les concentrations plasmatiques à létat déquilibre sont généralement atteintes en approximativement 15 jours avec des valeurs Cmax et ASC 4 à 7 fois plus élevées que celles observées après administration unique. A létat déquilibre, une valeur Cmax denviron 2 µg/ml est atteinte après administration orale de 200 mg par jour.
La demi-vie délimination terminale de litraconazole varie généralement de 16 à 28 heures après une administration unique et augmente pour atteindre 34 à 42 heures après administrations répétées. Lorsque le traitement est arrêté, les concentrations plasmatiques en itraconazole diminuent pour atteindre des concentrations presque indétectables en 7 jours à 14 jours, suivant la dose et de la durée du traitement.
La clairance plasmatique totale moyenne de litraconazole après administration intraveineuse est de 278 ml/min. La clairance de litraconazole diminue aux doses élevées, du fait dune saturation de son métabolisme hépatique.
Absorption
Litraconazole est rapidement résorbé après administration de la solution buvable. Le pic plasmatique de litraconazole est atteint environ 2,5 heures après administration à jeun de la solution buvable.
La biodisponibilité absolue observée de litraconazole administré en présence de nourriture est denviron 55% et augmente de 30% quand la solution buvable est administrée à jeun.
Lexposition à litraconazole est plus élevée avec la solution buvable quavec les gélules lorsque la même dose est administrée. (voir rubrique 4.4)
Dans certains groupes de patients, la biodisponibilité de litraconazole est diminuée dun facteur 1,5 à 2,5 (sujet HIV, neutropénique, mycose systémique à Aspergillus) pouvant atteindre 3 à 5 (dialyse péritonéale, mycose systémique à Cryptococcus). Lensemble de ces données se traduit par une très forte variabilité interindividuelle du paramètre biodisponibilité.
Distribution
La majorité de litraconazole est liée aux protéines plasmatiques (99,8%) et plus particulièrement à lalbumine (99,6% pour le métabolite hydroxylé). Il présente également une affinité pour les lipides. Seulement 0,2% de litraconazole plasmatique est libre.
Le volume de distribution est élevé (700 l) et indique une large pénétration tissulaire associée à un fort tropisme cellulaire.
Les concentrations dans les poumons, les reins, le foie, les os, lestomac, la rate et les muscles trouvées ont été deux à trois plus élevées que celles dans le plasma. La recapture par les tissus kératineux, et plus particulièrement par la peau est jusquà 4 fois plus importante que dans le plasma.
Les concentrations dans le liquide cérébrospinal sont bien plus faibles que dans le plasma.
Biotransformation
Litraconazole est métabolisé de façon extensive dans le foie en de nombreux métabolites. Les études in vitro ont montré que le CYP 3A4 est la principale enzyme impliquée dans le métabolisme de litraconazole.
Le métabolite principal est lhydroxy-itraconazole qui possède in vitro une activité antifongique comparable à celle de litraconazole. Les taux plasmatiques de ce métabolite sont environ 2 fois plus élevés que ceux de litraconazole.
Excrétion
Litraconazole est principalement éliminé sous forme de métabolites inactifs dans les urines (35%) et dans les fécès (54%) dans la semaine après ladministration dune dose sous forme de solution buvable. Lexcrétion rénale de litraconazole et de son métabolite actif, lhydroxy-itraconazole, représente moins de 1% de la dose intraveineuse administrée. Sur la base dune dose orale ditraconazole radio marquée, lexcrétion fécale du produit inchangé varie entre 3 et 18% de la dose administrée.
Populations particulières
Insuffisants hépatiques
Litraconazole est principalement métabolisé par le foie. Une étude pharmacocinétique a été conduite chez 6 sujets sains et 12 sujets cirrhotiques ayant reçu une seule dose ditraconazole (gélule de 100 mg). Une diminution statistiquement significative de la Cmax moyenne de litraconazole (47%) et un doublement de sa demi-vie délimination (37± 17 heures versus 16 ± 5 heures) ont été observés chez les sujets cirrhotiques comparativement aux sujets sains. Toutefois, lexposition globale à litraconazole, calculée sur la base de lASC était similaire chez les patients cirrhotiques et chez les sujets sains. Les données sur lutilisation à long terme de litraconazole chez des patients cirrhotiques ne sont pas disponibles (voir rubriques 4.2 et 4.4).
Insuffisants rénaux
Les données disponibles sur lutilisation de litraconazole par voie orale chez des patients insuffisants rénaux sont limitées. Une étude pharmacocinétique dans laquelle litraconazole a été administré à la dose unique de 200 mg (quatre gélules de 50 mg) a été conduite dans trois groupes de patients ayant une insuffisance rénale (urémie : n=7 ; hémodialyse : n=7 ; et dialyse péritonéale continue ambulatoire: n=5). Chez les patients urémiques avec une clairance de la créatinine moyenne de 13 ml/min x 1,73 m2, lexposition, calculée sur la base de lASC, était légèrement réduite comparée à celle de la population normale. Cette étude na pas montré deffet significatif de lhémodialyse ou de la dialyse péritonéale continue ambulatoire sur la pharmacocinétique de litraconazole (Tmax, Cmax et ASC0-8h). Les profils des concentrations plasmatiques en fonction du temps ont montré une large variation interindividuelle entre les trois groupes.
Après administration intraveineuse dune seule dose, les demi-vies délimination terminale moyennes de litraconazole chez les patients atteints dune insuffisance rénale légère (définie dans létude par une ClCr de 50-79 ml/min), modérée (définie dans létude par une ClCr de 20-49 ml/min), et sévère (définie dans létude par une ClCr < 20 ml/min) étaient similaires à celles des sujets sains (intervalle de 42-49 heures chez les patients insuffisants rénaux versus 48 heures chez les patients sains). Lexposition globale à litraconazole, calculée sur la base de lASC, diminuait dapproximativement 30% et 40 % chez les patients insuffisants rénaux modérés et sévères respectivement, comparée aux sujets avec une fonction rénale normale.
Les données sur lutilisation à long terme de litraconazole chez des patients insuffisants rénaux ne sont pas disponibles La dialyse na pas deffet sur la demi-vie ou la clairance de litraconazole ou de lhydroxy-itraconazole (voir rubriques 4.2 et 4.4).
Population pédiatrique
Deux études de pharmacocinétiques ont été conduites chez des enfants neutropéniques âgés de 6 mois à 14 ans. Lors de ces études, litraconazole en solution buvable a été administré à la dose de 5 mg/kg une ou deux fois par jour. Lexposition à litraconazole a été quelques peu plus élevée chez les enfants plus âgés (6 à 14 ans) comparés aux enfants plus jeunes. Chez tous les enfants, les concentrations plasmatiques efficaces ditraconazole ont été atteintes entre 3 et 5 jours après linstauration du traitement et ont été maintenues pendant toute la durée du traitement.
Hydroxypropyl-ß-Cyclodextrine
La biodisponibilité orale de lhydroxypropyl-β-cyclodextrine utilisée en tant quagent solubilisant est en moyenne plus faible que 0,5% et est similaire à celle de lhydroxypropyl-β-cyclodextrine seule. Cette faible biodisponibilité orale de lhydroxypropyl-β-cyclodextrine nest pas modifiée par la présence de nourriture et est similaire après administration unique et administrations répétées.
5.3. Données de sécurité préclinique
· Hydroxypropyl-β-cyclodextrine (HP-β-CD)
Chez la souris, le rat et le chien, les études de toxicité aiguë et de toxicité par administration réitérée montrent une importante marge de sécurité de l'HP-β-CD administrée par voies orale et intraveineuse. La majorité des effets observés sont de caractère adaptatif (modifications histologiques du tractus urinaire, ramollissement des fécès dû à une rétention osmotique aqueuse au niveau du gros intestin, activation du système phagocytaire mononucléaire) et montrent une réversibilité satisfaisante.
De légères modifications hépatiques ont été observées à des doses correspondant à 30 fois la dose d'HP-β-CD proposée en clinique. L'HP-β-CD n'a pas d'effets sur la fertilité, ni d'effets embryotoxiques ou tératogènes directs et est dépourvue d'effets mutagènes.
Dans l'étude de carcinogénèse chez le rat, une augmentation de l'incidence des néoplasmes du gros intestin (à 5000 mg/kg/jour) et du pancréas exocrine (à 500 mg/kg/jour) a été observée.
Chez le rat, le développement de tumeurs du pancréas est dû à l'effet mitogène de la cholécystokine.
Ce phénomène n'a pas été observé dans l'étude de cancérogénèse réalisée chez la souris, ni dans l'étude de toxicité à 12 mois chez le chien ou dans l'étude de 2 ans chez le singe cynomolgus femelle. La cholécystokine ne semble pas avoir d'effet mitogène chez l'homme.
Lorsque l'on tient compte des surfaces corporelles, l'exposition à l'HP-β-CD chez l'homme à la posologie initiale recommandée en clinique pour ce médicament correspond à environ 1,7 fois l'exposition observée à la dose la plus basse dans l'étude réalisée chez le rat.
Bien qu'hypothétique, la survenue d'une telle pathologie chez l'homme ne peut être totalement éliminée.
En conséquence, une surveillance de l'amylasémie chez les sujets traités à long terme (pendant plusieurs mois) paraît nécessaire.
· Itraconazole
L'itraconazole a été étudié dans une batterie standard de tests de sécurité non clinique.
Différentes études de toxicité aiguë, conduites par voie orale chez la souris, le rat, le cobaye et le chien, à des doses ditraconazole allant de 20 à 320 mg/kg ont mis en évidence des marges de sécurité importantes (allant de 8 à 38 fois la dose maximale recommandée chez lhomme en mg/m2).
Les études de toxicité sub-chronique, conduites par voie orale chez le rat et le chien, à des doses ditraconazole allant de 2,5 à 160 mg/kg/jour, ont révélé plusieurs organes /tissus cibles dont notamment le cortex surrénal, les ovaires, le foie et le système phagocytaire mononucléé. Des anomalies du métabolisme lipidique ont également été notées, se traduisant par des cellules xanthomateuses dans divers organes.
Aux doses de 40 et 80 mg/kg/jour chez le rat (2 et 4 fois la dose maximale recommandée chez lhomme en mg/m2), les analyses histologiques du cortex surrénal ont montré un gonflement réversible avec une hypertrophie cellulaire de la zone réticulée et fasciculée, parfois associée à un amincissement de la zone glomérulée. Au niveau hépatique, des altérations réversibles de type vacuolisation des hépatocytes, ont été observées aux doses de 40 et 160 mg/kg/jour. Les modifications histologiques observées au niveau du système phagocytaire mononucléé consistent en la présence de macrophages présentant un contenu protéique accru dans divers tissus parenchymateux (poumons, foie, tissus lymphoïdes).
Dans une étude de toxicité par administration répétée conduite chez le chien juvénile, une baisse de la densité minérale osseuse globale a été observée après administration chronique de doses de 30 mg/kg/jour (6 fois la dose maximale recommandée chez lhomme en mg/m2) d'itraconazole.
De même, dans plusieurs études de toxicité chronique conduites chez le rat, à des doses ditraconazole allant de 10 à 80 mg/kg, des anomalies osseuses ont été observées, se caractérisant par une diminution de lactivité des plaques osseuses, un amincissement du tissu osseux compact des os longs et une augmentation de la fragilité osseuse.
Cancérogénicité et mutagénicité
Litraconazole ne sest pas révélé génotoxique dans différents essais in vitro et in vivo.
Une étude de 23 mois conduite chez la souris na pas révélé de potentiel cancérogène de litraconazole jusquà la dose de 80 mg/kg/jour. Une autre étude de 24 mois conduite chez le rat na pas révélé de potentiel cancérogène de litraconazole jusquà la dose de 20 mg/kg/jour ; cependant à la dose la plus forte testée (80 mg/kg/jour), une augmentation de lincidence de sarcome des tissus mous a été observée chez les rats mâles, pouvant possiblement être attribuée à des réactions inflammatoires, secondaires à une accumulation de cholestérol dans le tissu conjonctif.
Toxicologie de la reproduction
Chez le rat, l'itraconazole entraîne à des doses allant de 40 à 160 mg/kg/jour (2 à 10 fois la dose maximale recommandée chez lhomme en mg/m²), une augmentation dose-dépendante de la toxicité maternelle, de l'embryotoxicité et de la tératogénicité. Chez la souris, les mêmes observations sont faites pour des doses de 80 mg/kg/jour (2 fois la dose maximale recommandée chez lhomme en mg/m²).
Chez le rat, des anomalies majeures du squelette ont été observées dans la descendance ; et chez la souris, des encéphalocèles et des macroglossies.
Aucun effet tératogène na été mis en évidence chez le lapin jusquà une dose de 80 mg/kg (9 fois la dose maximale recommandée chez lhomme en mg/m2).
Fertilité
Les données animales disponibles chez le rat nont pas mis en évidence deffet délétère de l'itraconazole sur la fertilité mâle ou femelle (voir rubrique 4.6).
2 ans.
Après première ouverture du flacon, ce médicament doit être conservé maximum un mois.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
6.5. Nature et contenu de l'emballage extérieur
150 ml en flacon (verre brun) avec fermeture de sécurité enfant (polypropylène) et joint (PE) avec gobelet doseur gradué (polypropylène).
6.6. Précautions particulières délimination et de manipulation
SPORANOX solution buvable est fourni en flacon pourvu d'un bouchon avec fermeture de sécurité enfant. Pour l'ouvrir : appuyer sur le bouchon de plastique tout en le dévissant dans le sens inverse des aiguilles d'une montre.
Un gobelet-doseur gradué est fourni avec SPORANOX solution buvable. Utiliser ce gobelet-doseur dans le sens où il est posé sur le flacon. Sassurer que le côté avec les graduations (le côté avec le plus petit contenant) soit en haut. Si la flèche sur le côté pointe vers le haut, le gobelet-doseur est dans le bon sens.
Verser la solution buvable jusquà la graduation correspondant à la quantité à administrer prescrite.
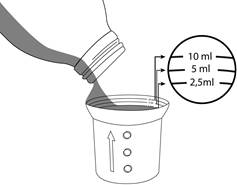
Après chaque usage, laver le gobelet-doseur soigneusement à l'eau tiède.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
1, RUE CAMILLE DESMOULINS
TSA 91003
92787 ISSY LES MOULINEAUX CEDEX 9
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 345 020 6 4: 150 ml en flacon (verre brun) avec gobelet-doseur gradué (polypropylène).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Prescription initiale hospitalière annuelle.