|
|
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 10/10/2019
PROLASTIN 1000 mg, poudre et solvant pour solution injectable / perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
1 flacon contient:1000 mg d'alpha-1 antitrypsine humaine
1 ml de solution reconstituée contient 25 mg dalpha-1 antitrypsine humaine
Excipient à effet notoire: la poudre pour solution injectable / perfusion contient 4,8 mmol (ou 110,35 mg) de sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution injectable / perfusion.
Poudre : blanche à beige.
Solvant: solution limpide et incolore.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Adultes, y compris personnes âgées
Sauf prescription contraire, la dose hebdomadaire est de 60 mg de principe actif par kg de poids corporel (équivalent à 180 ml de solution injectable / perfusion reconstituée, qui contient 25 mg/ml d'alpha-1 antitrypsine humaine pour un patient pesant 75 kg), administrée sous la forme dune perfusion de courte durée qui suffit habituellement pour maintenir un taux d'alpha-1 antitrypsine sérique constant supérieur à 80 mg/dl correspondant à un taux de 1,3 μM au niveau des poumons. Ces taux dans le sang et dans le liquide recouvrant lépithélium pulmonaire sont, en théorie, censés protéger contre l'aggravation de l'emphysème pulmonaire.
Population pédiatrique
Aucune donnée nest disponible concernant lutilisation de PROLASTIN chez les enfants et adolescents âgés de 0 à 18 ans.
Mode dadministration
La substance sèche doit être mélangée au solvant (40 ml d'eau pour préparations injectables) et dissoute comme décrit dans la rubrique 6.6.
La solution reconstituée apparaît comme une solution claire à opalescente, incolore ou dune couleur jaunâtre à verte. La solution reconstituée doit être administrée en perfusion intraveineuse lente, en utilisant un set pour perfusion approprié. La vitesse de perfusion ne peut pas dépasser 0,08 ml/kg de poids par minute (correspond à 6 ml par minute pour un patient de 75 kg).
La solution préparée doit être administrée dans les 3 heures qui suivent sa préparation.
La durée du traitement est laissée à l'appréciation du médecin traitant. Aucune limite de durée de traitement spécifique na été fixée.
Le traitement ou la surveillance du traitement des patients présentant un déficit en alpha-1 antitrypsine doivent être confiés à des médecins expérimentés dans le traitement les broncho-pneumopathies chroniques obstructives.
PROLASTIN ne doit pas être utilisé chez les patients :
· souffrant d'un déficit en IgA sélectif, chez qui la présence d'anticorps anti-IgA a été démontrée, en raison du risque de réactions allergiques voire de choc anaphylactiques.
· ayant une hypersensibilité à lalpha-1 antitrypsine ou à lun des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Étant donné que PROLASTIN peut provoquer une augmentation passagère du volume sanguin, la prudence simpose plus particulièrement chez les patients présentant une insuffisance cardiaque sévère et les patients présentant un risque de surcharge volémique.
Les mesures standard destinées à prévenir les infections provoquées par l'utilisation des médicaments préparés à partir de sang ou de plasma humains, comprennent la sélection des donneurs, lanalyse des dons de sang individuels et des pools plasmatiques au niveau de certains marqueurs infectieux spécifiques ainsi que la mise en uvre dans le procédé de fabrication, détapes efficaces d'inactivation / élimination des virus. Malgré ces mesures, lorsque des médicaments préparés à partir de sang ou de plasma humains sont administrés, le risque de transmission dagents infectieux ne peut pas être totalement exclu. Ceci s'applique également aux virus inconnus ou émergents et à d'autres agents pathogènes.
Les mesures prises sont considérées comme efficaces contre les virus enveloppés, notamment le virus de limmunodéficience humaine (VIH), le virus de lhépatite B (VHB) et de lhépatite C (VHC). Les mesures prises peuvent avoir un effet limité contre les virus non enveloppés tels que l'hépatite A et le parvovirus B19. L'infection par le parvovirus B19 peut être grave chez la femme enceinte (infection ftale) ainsi que chez les patients immunodéprimés ou ayant une augmentation de lérythropoïèse (par exemple en cas danémie hémolytique).
Une vaccination appropriée (hépatite A et B) des patients recevant régulièrement ou de façon répétée de lalpha-1 antitrypsine préparé à partir de plasma humain est recommandée.
Il est fortement recommandé que chaque fois que PROLASTIN est administré à un patient, le nom et le numéro de lot du produit soient enregistrés afin de conserver un lien entre le patient et le lot du produit.
PROLASTIN contient 4,8 mmol de sodium par flacon (ce qui correspond à une dose de 21,6 mmol de sodium dans le cas d'un patient de 75 kg). Ceci doit être pris en compte chez les patients qui doivent suivre un régime pauvre en sodium.
Le traitement de PROLASTIN ne peut pas être refusé aux fumeurs. Mais étant donné que l'efficacité de PROLASTIN peut être compromise par la présence de fumée dans les poumons, il est fortement recommandé à ces patients d'arrêter de fumer.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction entre PROLASTIN et d'autres médicaments n'est connue à ce jour
4.6. Fertilité, grossesse et allaitement
Grossesse
On ne dispose d'aucune donnée clinique sur l'exposition à PROLASTIN pendant la grossesse. Aucune étude sur l'animal n'a été réalisée dans ce domaine. Lors de la prescription de PROLASTIN à des femmes enceintes, la prudence simpose.
On ne sait pas si lalpha-1 antitrypsine est excrétée dans le lait maternel humain. Le passage de l alpha-1 antitrypsine dans le lait n'a pas été étudié chez l'animal. La décision de poursuivre / d'interrompre l'allaitement ou de poursuivre / d'arrêter le traitement par PROLASTIN doit être prise en tenant compte de l'avantage de l'allaitement pour l'enfant et du bénéfice du traitement par PROLASTIN pour la femme.
Fertilité
Sans objet.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
PROLASTIN na aucun effet sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les effets indésirables suivants ont été observés dans le cadre de l'utilisation de PROLASTIN :
|
Classe de systèmes dorganes |
Peu fréquents |
Rares |
Très rares |
|
Affections cardiaques |
|
Tachycardie |
|
|
Troubles généraux et anomalies au niveau du site d'administration |
Frissons, fièvre, symptômes pseudo-grippaux, douleur à la poitrine |
|
|
|
Affections du système immunitaire |
Urticaire |
Réactions d'hypersensibilité |
Choc anaphylactique |
|
Affections du système nerveux |
Vertiges / confusion / céphalées |
|
|
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée |
|
|
|
Affections de la peau et du tissu sous-cutané |
Éruption cutanée
|
|
|
|
Affections vasculaires |
|
Hypotension |
|
|
Affections gastro-intestinales |
Nausées |
|
|
|
Affections musculo-squelettiques et systémiques |
Douleur articulaire / arthralgie |
Dorsalgies |
|
Le traitement par PROLASTIN peut provoquer des réactions connues notamment de la fièvre, des symptômes pseudo-grippaux, une dyspnée, de l'urticaire, des nausées, etc. Comme avec tout traitement protéique, des réactions immunologiques peu fréquentes ou rares peuvent être observées. Il peut s'agir de réactions allergiques telles que : urticaire, dyspnée, arthralgie et très rarement choc anaphylactique. Les symptômes pouvant être de nature immunologique doivent être évalués avant que le patient ne reprenne son traitement.
En ce qui concerne la sécurité virale, voir rubrique 4.4.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Les conséquences du surdosage ne sont pas connues.
En cas de surdosage, le patient doit être étroitement surveillé pour détecter d'éventuels effets indésirables et les mesures dintervention nécessaires doivent être disponibles.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
L'alpha-1 antitrypsine est un composant normal du sang humain qui inhibe l'activité de l'élastase du neutrophile et d'autres enzymes. L'alpha-1 antitrypsine a un poids moléculaire de 51 kDa et appartient à la famille des inhibiteurs de la sérine-protéase.
Actuellement, on suppose que la pathogenèse de l'emphysème lié à un déficit en alpha-1 antitrypsine est attribuable à la perturbation biochimique chronique de l'équilibre entre l'élastase et lalpha-1 antitrypsine. L'élastase, qui est synthétisée par les cellules pro-inflammatoires dans les voies respiratoires inférieures, est capable de dégrader les tissus élastiques. Un des principaux inhibiteurs de l'élastase est l'alpha-1 antitrypsine, absent en cas de déficit congénital en alpha-1 antitrypsine. Dans ce cas, les structures alvéolaires ne sont pas protégées contre l'élastase qui est libérée par les neutrophiles dans les voies respiratoires inférieures, à laquelle elles sont donc exposées de manière chronique.
Cette situation entraîne une dégradation progressive des tissus élastiques et lorsque les taux d'alpha-1 antitrypsine baissent en dessous de 80 mg/dl, le risque d'emphysème augmente.
Deux études observationnelles contrôlées ont montré que le ralentissement le plus significatif de la réduction du VEMS était celui observé chez les patients ayant un VEMS de 35 à 60% de la valeur attendue.
5.2. Propriétés pharmacocinétiques
5.3. Données de sécurité préclinique
Le principe actif de PROLASTIN, l'alpha-1 antitrypsine, est obtenu à partir de plasma humain et se comporte comme un composant plasmatique endogène. L'administration d'une dose unique de PROLASTIN à différentes espèces animales aussi bien que l'administration de doses quotidiennes pendant 5 jours consécutifs à des lapins n'ont montré aucun effet toxique. On ne dispose pas d'études précliniques complémentaires sur l'administration de doses répétées (toxicité chronique, carcinogénicité, reprotoxicité). Il ny aurait aucun intérêt à mener ces études sur les modèles animaux traditionnels étant donné quil est fort probable que les animaux développeraient des anticorps contre les protéines hétérologues humaines administrées.
Poudre : chlorure de sodium, dihydrophosphate de sodium.
Solvant : eau pour préparations injectables.
2 ans.
La solution préparée doit être utilisée dans les 3 heures suivant sa préparation.
6.4. Précautions particulières de conservation
À conserver à une température ne dépassant pas 25°C. Ne pas congeler.
Une fois préparée, la solution injectable / perfusion ne peut plus être conservée au réfrigérateur.
Éliminez la solution inutilisée conformément à la réglementation locale en vigueur.
6.5. Nature et contenu de l'emballage extérieur
Poudre : flacon en verre de type I avec bouchon en caoutchouc isoprène et capsule en aluminium.
Solvant : flacon en verre de type I avec bouchon en caoutchouc chlorobutyle et dune capsule en aluminium.
Chaque emballage d'origine contient :
· un flacon de poudre 1000 mg d'alpha-1 antitrypsine humaine ;
· un flacon de solvant (40 ml deau pour préparations injectables) ;
· un dispositif de transfert Mix2Vial pour la reconstitution.
6.6. Précautions particulières délimination et de manipulation
La substance sèche doit être mélangée et dissoute dans le contenu du flacon de 40 ml d'eau pour préparations injectables, comme décrit ci-dessous. La solution reconstituée apparaît comme une solution claire à opalescente, incolore ou dune couleur verte à jaunâtre. La reconstitution totale doit être obtenue en 5 minutes.
Préparation de la solution reconstituée pour perfusion
1. Pendant la procédure de reconstitution, utilisez une technique aseptique (matériel propre et stérilisé) et une surface de travail plane.
2. Avant lutilisation, amenez les flacons de PROLASTIN et de solvant (eau stérile pour préparations injectables) à température ambiante (20-25ºC).
3. Enlevez la capsule protectrice du flacon de PROLASTIN et nettoyez le dessus du bouchon avec un coton imbibé dalcool. Laissez sécher le bouchon en caoutchouc.
4. Répétez cette étape avec le flacon deau stérile.
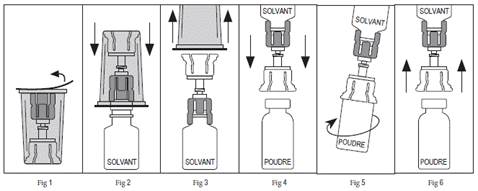
5. Ouvrez lemballage du dispositif stérile Mix2Vial en détachant le feuillet (Figure 1). Nenlevez pas le dispositif de lemballage.
6. Placez le flacon de solvant bien droit sur une surface plane. En tenant bien fermement le flacon de solvant, enfoncez verticalement lextrémité bleue du dispositif Mix2Vial jusquà ce que la pointe pénètre dans le bouchon (Figure 2).
7. Enlevez lemballage extérieur transparent du dispositif Mix2Vial et jetez-le (Figure 3).
8. Placez le flacon de PROLASTIN bien droit sur une surface plane et retournez le flacon de solvant avec le dispositif Mix2Vial toujours attaché.
9. En tenant bien fermement le flacon de PROLASTIN sur une surface plane, enfoncez verticalement lextrémité transparente du dispositif Mix2Vial jusquà ce que la pointe pénètre dans le bouchon (Figure 4). Le solvant sera automatiquement transféré dans le flacon de PROLASTIN car il est sous vide.
Remarque : si le dispositif Mix2Vial est connecté sous un angle, le vide peut ne pas se maintenir dans le flacon du produit et le solvant ne sera pas automatiquement transféré dans le flacon du produit. Si le flacon nest plus sous vide, utilisez une seringue et une aiguille stériles pour enlever leau stérile du flacon de solvant et linjecter dans le flacon de PROLASTIN, en dirigeant le jet de liquide le long de la paroi du flacon.
10. Tout en laissant les flacons de solvant et de PROLASTIN attachés au dispositif Mix2Vial, remuez doucement (Figure 5) jusquà la dissolution complète de la poudre. Ne secouez pas afin déviter la formation de mousse. La solution reconstituée doit être limpide. Ne lutilisez pas si elle contient des particules ou si elle présente une coloration anormale.
11. Étant donné quil est nécessaire dutiliser plus dun flacon pour obtenir la dose requise, répétez les instructions ci-dessus en utilisant un autre emballage contenant un nouveau dispositif Mix2Vial. Ne réutilisez pas le même Mix2Vial.
12. Enlevez le dispositif Mix2Vial (Figure 6) et administrez le produit en utilisant une technique aseptique.
|
|
Seules les solutions claires peuvent être utilisées. La solution reconstituée doit toujours être utilisée dans les 3 heures qui suivent sa reconstitution. Les solutions non utilisées doivent être éliminées.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
COLMARER STRASSE 22
60528 FRANKFURT
ALLEMAGNE
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 301 883 1 6 : Poudre en flacon (verre) et solvant en flacon (verre) avec dispositif de transfert.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière.