RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 27/10/2020
1. DENOMINATION DU MEDICAMENT
BERINERT 2000 UI, poudre et solvant pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Substance active : Inhibiteur de la C1 estérase humaine, sous cutanée (s.c.)
BERINERT 2000 UI contient 2000 UI dinhibiteur de la C1 estérase par flacon à injecter.
Lactivité en inhibiteur de la C1 estérase est exprimée en Unité Internationale (UI), conformément au standard actuel de lOMS pour les inhibiteurs de la C1 estérase.
BERINERT 2000 UI contient 500 UI/ml dinhibiteur de la C1 estérase après reconstitution avec 4 ml d'eau pour préparations injectables.
La teneur en protéines totales de la solution reconstituée est de 65 mg/ml.
Excipient(s) à effet notoire :
Sodium jusqu'à 486 mg (approximativement 21 mmol) pour 100 ml de solution.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Poudre et solvant pour solution injectable
Poudre blanche
4. DONNEES CLINIQUES
4.1. Indications thérapeutiques
BERINERT pour injection sous cutanée est indiqué dans la prévention des crises récidivantes dangidème héréditaire (AOH) chez les patients adolescents et adultes présentant un déficit en inhibiteur de la C1 estérase.
4.2. Posologie et mode d'administration
BERINERT est destiné à l'auto-administration par injection sous-cutanée. Le patient ou lentourage soignant doit être formé sur la manière d'administrer BERINERT le cas échéant.
Posologie
La dose recommandée de BERINERT SC est de 60 UI/kg de poids corporel, deux fois par semaine (tous les 3-4 jours)
Population pédiatrique
La posologie chez les adolescents est la même que chez les adultes.
Mode dadministration
Ladministration se fait uniquement par voie sous-cutanée.
Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Il est recommandé de réaliser linjection sous-cutanée de BERINERT dans la région abdominale. Au cours des essais cliniques, un seul site d'injection a été utilisé pour ladministration de BERINERT.
La préparation reconstituée doit être administrée par injection sous-cutanée à un débit toléré par le patient.
4.3. Contre-indications
Ce médicament est contre-indiqué chez les patients qui ont développé des réactions dhypersensibilité, y compris une anaphylaxie, engageant le pronostic vital aux préparations à base dinhibiteur de la C1 estérase ou à lun des excipients mentionnés dans la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Réactions dhypersensibilité
En cas de survenue d'une réaction allergique dintensité sévère, il convient d'arrêter immédiatement ladministration de BERINERT (par exemple, en arrêtant linjection) et d'instaurer un traitement médical approprié.
En cas de crise dAOH aiguë, un traitement personnalisé doit être instauré.
Évènements thromboemboliques (ETE)
Une thrombose est survenue lors dessais de traitement avec de fortes doses dinhibiteur de la C1 estérase administrées par voie I.V. dans le cadre de la prophylaxie ou du traitement du syndrome de fuite capillaire, avant, pendant ou après une chirurgie cardiaque réalisée sous circulation extracorporelle (utilisation hors indication et doses autorisées). Lexistence dun lien de causalité entre ladministration par voie S.C. dune solution dinhibiteur de la C1 estérase aux doses recommandées et la survenue dun ETE na pas pu être établie.
Sécurité virale
Les mesures habituelles de prévention du risque de transmission dagents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, la recherche des marqueurs spécifiques dinfection sur chaque don et sur les mélanges de plasma ainsi que la mise en uvre dans le procédé de fabrication détapes efficaces pour linactivation/élimination virale. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission dagents infectieux ne peut pas être totalement exclu. Ceci sapplique également aux virus inconnus ou émergents et aux autres agents pathogènes.
Les mesures prises sont considérées comme efficaces vis à vis des virus enveloppés tels que le virus de limmunodéficience humaine (VIH), le virus de lhépatite B (VHB), le virus de lhépatite C (VHC) et vis-à-vis des virus non enveloppés tels que le VHA et le parvovirus B19.Une vaccination appropriée (hépatite A et B) doit être envisagée chez les patients qui reçoivent régulièrement ou à plusieurs reprises des produits dérivés du plasma humain.
BERINERT 2000 UI contient moins de 1 mmol de sodium (23 mg) par flacon, cest-à-dire quil est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude dinteraction na été réalisée.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il existe des données limitées suggérant labsence daugmentation du risque lors de l'utilisation de produits à base dinhibiteur de la C1 estérase humaine chez la femme enceinte. Linhibiteur de la C1 estérase est un composant physiologique du plasma humain. Aucune étude de toxicité sur la reproduction et sur le développement na été réalisée avec BERINERT chez les animaux. Aucun effet indésirable sur la fertilité ou sur le développement pré et post-natal n'est attendu chez l'Homme.
Dans trois études, ayant inclus 344 patients, les données de 36 femmes (50 grossesses) ont été recueillies et aucun événement indésirable na été associé au traitement par inhibiteur de la C1 estérase avant, pendant ou après la grossesse, et les femmes ont accouché de bébés en bonne santé.
Allaitement
Il ny a pas de donnée sur lexcrétion de BERINERT dans le lait maternel, leffet sur le nourrisson allaité, ou les effets sur la production de lait. Il convient de considérer les bénéfices de lallaitement pour le développement et la santé du nourrisson, conjointement avec le besoin clinique de la mère concernant BERINERT et les éventuels effets indésirables potentiels de BERINERT sur le nourrisson allaité ou de laffection sous-jacente de la mère.
Fertilité
Linhibiteur de la C1 estérase est un composant physiologique du plasma humain. Aucune étude sur la reproduction ou sur la toxicité liée au développement na été menée chez lanimal avec BERINERT.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
BERINERT na aucun effet ou un effet négligeable sur laptitude à conduire des véhicules et à utiliser des machines.
4.8. Effets indésirables
Les réactions indésirables ont été recueillies à partir de lÉtude 3001, une étude pivot de phase 3 qui a été menée chez des patients (n = 86) atteints dAOH et recevant BERINERT par voie sous-cutanée. Les patients éligibles pouvaient également participer à une étude dextension en ouvert (Étude 3002) pendant une durée allant jusquà 140 semaines (n = 126). La fréquence des réactions indésirables sappuie sur les événements qui sont associés à ladministration de BERINERT. Elle a été estimée en fonction de chaque patient et classée de la manière suivante :
Très fréquent : (≥ 1/10)
Fréquent : (≥ 1/100 et < 1/10)
Peu fréquent : (≥ 1/1 000 et < 1/100)
Rare : (≥ 1/10 000 et < 1/1 000)
Très rare : (< 1/10 000)
|
Classification MedDRA par Classe de Systèmes dOrganes
|
Réaction indésirable
|
Fréquence
|
|
Infections et infestations
|
Rhinopharyngite
|
Très fréquent
|
|
Affections du système immunitaire
|
Hypersensibilité
(Hypersensibilité, prurit, éruption cutanée et urticaire)
|
Fréquent
|
|
Affections du système nerveux
|
Vertiges
|
Fréquent
|
|
Troubles généraux et anomalies au site dadministration
|
Réactions au site dinjectiona
|
Très fréquent
|
|
a Contusion au site dinjection, Sensation de froid au site dinjection, Écoulement au site dinjection, Érythème au site dinjection, Hématome au site dinjection, Hémorragie au site dinjection, Induration au site dinjection, dème au site dinjection, Douleur au site dinjection, Prurit au site dinjection, Éruption cutanée au site dinjection, Réaction au site dinjection, Cicatrice au site dinjection, Gonflement au site dinjection, Urticaire au site dinjection, Sensation de chaleur au site dinjection.
|
Population pédiatrique
Le profil de sécurité de BERINERT a été évalué dans un sous-groupe de onze patients âgés de 8 à < 17 ans, dans les deux études (Étude 3001, Étude 3002) et était cohérent avec les résultats globaux de sécurité.
Autres populations spéciales
Population âgée
Le profil de sécurité de BERINERT a été évalué dans un sous-groupe de dix patients âgés de 65 à 72 ans, dans les deux études (Étude 3001, Étude 3002) et était cohérent avec les résultats globaux de sécurité.
Pour la sécurité vis-à-vis des agents transmissibles, voir la rubrique 4.4.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
4.9. Surdosage
Aucun cas de surdosage n'a été rapporté. Dans une étude clinique à doses fixes, des doses allant jusquà 117 UI/kg ont été administrées par voie S.C. deux fois par semaine et elles ont été bien tolérées par les patients.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Autres agents hématologiques, médicaments utilisés dans le traitement de langidème héréditaire : Inhibiteur de la C1 estérase, dérivés du plasma, code ATC : B06AC01
L'inhibiteur de la C1 estérase est une glycoprotéine de plasma dont le poids moléculaire est égal à 105 kD et le taux d'hydrate de carbone à 40 %. Sa concentration dans le plasma humain est denviron 240 mg/l. En plus du plasma humain, le placenta, les cellules hépatiques, les monocytes et les plaquettes contiennent également de l'inhibiteur de la C1 estérase.
L'inhibiteur de la C1 estérase appartient au système inhibiteur de protéase à sérine (serpine) du plasma humain, tout comme d'autres protéines : antithrombine III, alpha-2 antiplasmine, alpha-1-antitrypsine et dautres.
Mécanisme daction
Dans les conditions physiologiques, linhibiteur de la C1 estérase inhibe la voie classique du système du complément en inactivant les composants enzymatiques actifs C1s et C1r. Lenzyme active et linhibiteur forment un complexe stchiométrique 1:1.
Par ailleurs, linhibiteur de la C1 estérase représente le plus important inhibiteur dans lactivation de la phase contact de la coagulation en inhibant le facteur XIIa et ses fragments. De plus, il est également le principal inhibiteur, avec la macroglobuline alpha-2, de la kallicréine plasmatique.
L'effet thérapeutique de BERINERT dans le traitement de l'angidème héréditaire est induit par la substitution de l'activité déficiente de l'inhibiteur de la C1 estérase.
Efficacité et sécurité clinique
Lefficacité et la sécurité de BERINERT en prophylaxie de routine pour prévenir les crises dAOH ont été démontrées dans une étude multicentrique, randomisée, en double aveugle, contrôlée et croisée versus placebo (Étude 3001). Létude a évalué 90 patients adultes et adolescents présentant un AOH symptomatique de type I ou II. Lâge médian (plage) des patients était de 40 ans (de 12 à 72 ans) ; 60 patients étaient des femmes et 30 étaient des hommes. Les patients ont été randomisés pour recevoir soit 60 UI/kg, soit 40 UI/kg, de BERINERT sur une période de traitement de 16 semaines et un placebo sur lautre période de traitement de 16 semaines. BERINERT ou le placebo a été administré par les patients eux-mêmes par voie sous-cutanée deux fois par semaine. Lefficacité a été évaluée pendant les 14 dernières semaines de chaque période de traitement. Les patients éligibles pouvaient également participer à une étude dextension en ouvert de 140 semaines au maximum (Étude 3002). Environ la moitié des patients recrutés dans létude dextension avaient participé à lÉtude 3001 (64/126, 50,8 %), ce qui a contribué aux similitudes entre les populations détude.
Étude 3001 :
Les doses S.C. de 60 UI/kg ou de 40 UI/kg de BERINERT deux fois par semaine ont conduit à une différence significative du nombre de crises dAOH normalisées en fonction du temps (taux de crises) par rapport au placebo (Tableau 1). Le nombre de crises dAOH normalisé en fonction du temps chez les patients ayant reçu 60 UI/kg était de 0,52 crises par mois, contre 4,03 crises par mois avec le placebo (p < 0,001). Le nombre de crises dAOH normalisé en fonction du temps chez les patients ayant reçu 40 UI/kg était de 1,19 crises par mois, contre 3,61 crises par mois avec le placebo (p < 0,001).
Tableau 1 : Nombre de crises dAOH normalisé en fonction du temps (Nombre/Mois)
|
|
60 UI/kg
Séquences de traitement
(N = 45)
|
40 UI/kg
Séquences de traitement
(N = 45)
|
|
PRODUIT
|
Placebo
|
PRODUIT
|
Placebo
|
|
n
|
43
|
42
|
43
|
44
|
|
Moyenne (ET)
|
0,5 (0,8)
|
4,0 (2,3)
|
1,2 (2,3)
|
3,6 (2,1)
|
|
Mini, Maxi
|
0,0, 3,1
|
0,6, 11,3
|
0,0, 12,5
|
0,0, 8,9
|
|
Médiane
|
0,3
|
3,8
|
0,3
|
3,8
|
|
Moyenne des MC (ES)*
|
0,5 (0,3)
|
4,0 (0,3)
|
1,2 (0,3)
|
3,6 (0,3)
|
|
IC à 95 % de la moyenne de MC*
|
(0,0, 1,0)
|
(3,5, 4,6)
|
(0,5, 1,9)
|
(3, 4,3)
|
|
Différence entre traitements
(intra-patients)
|
60 UI/kg Placebo
|
40 UI/kg Placebo
|
|
Moyennes des MC* (IC à 95 %)
|
‑3,5 (‑4,2, ‑2,8)
|
‑2,4 (‑3,4, ‑1,5)
|
|
Valeur p*
|
< 0,001
|
< 0,001
|
IC = intervalle de confiance ; AOH = angidème héréditaire ; N = nombre de sujets randomisés ; n = nombre de sujets avec des données ; MC = moindres carrés.
* Daprès un modèle mixte.
Le pourcentage médian (25e, 75e percentiles) de réduction du nombre de crises dAOH normalisé en fonction du temps par rapport au placebo était de 95 % (79, 100) à la dose de 60 UI/kg et de 89 % (70, 100) à la dose de 40 UI/kg de BERINERT chez les sujets ayant des données évaluables dans les deux périodes de traitement.
Le pourcentage de répondeurs (IC à 95 %) présentant une réduction ≥ 50 % du nombre de crises dAOH normalisé en fonction du temps sous BERINERT par rapport au placebo était de 83 % (73 %, 90 %). Quatre-vingt-dix pour cent (90 %) des patients recevant la dose de 60 UI/kg ont répondu au traitement et 76 % des patients recevant la dose de 40 UI/kg ont répondu au traitement.
Soixante-et-onze pour cent (71 %) des patients recevant la dose de 60 UI/kg et 53 % des patients recevant la dose de 40 UI/kg ont présenté ≥1 crise dAOH par période de 4 semaines sous placebo et <1 crise dAOH par période de 4 semaines sous BERINERT.
Un total de 40 % des patients recevant la dose de 60 UI/kg et de 38 % des patients recevant la dose de 40 UI/kg na pas présenté de crise et le taux médian de crises dAOH par mois était de 0,3 avec les deux doses.
BERINERT a conduit à une différence significative du nombre dutilisations de médicaments de secours (taux dutilisation de médicaments de secours) normalisé en fonction du temps par rapport au placebo. Une dose de 60 UI/kg a conduit à un taux moyen dutilisation de médicament de secours de 0,3 utilisations par mois, contre 3,9 utilisations par mois avec le placebo. Une dose de 40 UI/kg a conduit à un taux moyen dutilisation de médicament de secours de 1,1 utilisation par mois, contre 5,6 utilisations par mois avec le placebo.
Étude 3002 :
La sécurité et lefficacité à long terme de BERINERT en prophylaxie de routine pour prévenir les crises dAOH ont été démontrées dans une étude randomisée, en ouvert, avec bras parallèles. Létude a évalué 126 patients adultes et pédiatriques présentant un AOH symptomatique de type I ou II, dont 64 patients étaient inclus de létude 3001 et 62 étaient des nouveaux patients. Lâge médian (plage) des patients était de 41,0 (8-72) ans. Des patients présentant un taux mensuel de crises de 4,3 sur 3 mois avant linclusion dans létude ont été recrutés et traités pendant une moyenne de 1,5 ans ; 44 patients (34,9 %) ont été exposés pendant plus de 2 ans. Lactivité fonctionnelle moyenne de linhibiteur de la C1 estérase à léquilibre a augmenté à 52,0 % avec la dose de 40 UI/kg et à 66,6 % avec la dose de 60 UI/kg. Lincidence des événements indésirables était faible et similaire dans les deux groupes de dose (respectivement 11,3 et 8,5 évènements par année-patient pour 40 UI/kg et 60 UI/kg).
La moyenne (ET) du nombre de crises dAOH normalisé en fonction du temps était de 0,45 (0,737) crises par mois pour 40 UI et de 0,45 (0,858) crises par mois pour 60 UI.
Le pourcentage de répondeurs (IC à 95%) présentant une réduction ≥ 50 % du nombre de crises dAOH normalisé en fonction du temps sous BERINERT par rapport au nombre de crises dAOH normalisé en fonction du temps utilisé pour qualifier la participation à l'Étude 3002 était de 93,5 % (84,6 %, 97,5 %) dans le groupe de traitement de 40 UI/kg et de 91,7 % (81,9 %, 96,4 %) dans le groupe de traitement de 60 UI/kg.
Le pourcentage de sujets présentant une fréquence de crise d'AOH normalisée en fonction du temps de < 1 crise d'AOH par période de 4 semaines était de 79,4 % pour 40 UI/kg et de 85,7 % pour 60 UI/kg.
Le pourcentage de sujets sans crise d'AOH était de 34,9 % pour 40 UI/kg et de 44,4 % pour 60 UI/kg (pendant toute la durée de l'étude avec une durée maximale d'exposition de > 2,5 ans). Sur les 23 patients ayant reçu 60 UI/kg pendant plus de 2 ans, 19 (83 %) n'ont présenté aucune crise pendant les mois 25 à 30 du traitement.
Le nombre moyen normalisé dans le temps d'utilisations de médicaments de secours était de 0,26 (0,572) utilisation par mois pour 40 UI/kg et de 0,31 (0,804) utilisations par mois pour 60 UI/kg.
Population pédiatrique
La sécurité et l'efficacité de BERINERT ont été évaluées dans un sous-groupe de 11 patients âgés de 8 à < 17 ans dans le cadre dune étude de prophylaxie en routine randomisée, en double aveugle, contrôlée et croisée versus placebo (Étude 3001) et d'une étude randomisée, ouverte, contrôlée versus traitement actif (Étude 3002). Les résultats de l'analyse de sous-groupe par âge étaient cohérents avec les résultats globaux de l'étude.
5.2. Propriétés pharmacocinétiques
Les caractéristiques pharmacocinétiques (PK) de BERINERT sous-cutanée ont principalement été décrites à laide de méthodes de PK de population appliquées sur des données regroupées issues de 3 essais cliniques menés auprès de sujets sains et de sujets présentant un AOH.
Absorption
Après administration par voie sous-cutanée deux fois par semaine, BERINERT est absorbé lentement ; le temps médian (IC à 95 %) écoulé avant lobtention de la concentration maximale (tmax) étant denviron 59 heures (23 à 134 heures). Daprès la demi-vie plasmatique apparente médiane (IC à 95 %) de 69 heures (24 à 250 heures), létat déquilibre de l'inhibiteur de la C1 estérase devrait être obtenu au bout de 3 semaines dadministration. Létat déquilibre fonctionnel minimal moyen (IC à 95 %) de l'inhibiteur de la C1 estérase devrait atteindre un taux de 48 % (25,1 à 102 %) suite à une administration de BERINERT par voie S.C. deux fois par semaine à une dose de 60 UI/kg. La biodisponibilité relative (F) moyenne (IC à 95 %) de BERINERT après une administration S.C. était estimée à environ 43 % (35,2 à 50,2 %).
Distribution et Élimination
Dans la population, la clairance moyenne (IC à 95 %) et le volume de distribution apparent de BERINERT étaient estimés à environ 83 ml/h (72,7 à 94,2 ml/h) et 4,33 l (3,5 à 5,15 l), respectivement. Il a été observé que la clairance de l'inhibiteur de la C1 estérase était directement proportionnelle au poids corporel total. À létat déquilibre, il a été observé que les paramètres PK de BERINERT en administration S.C. étaient indépendants de la dose chez les patients présentant un AOH qui recevaient des doses se situant entre 20 et 80 UI/kg.
Les paramètres PK de l'inhibiteur de la C1 estérase nont fait lobjet daucune étude menée au sein de populations particulières de patients stratifiées selon le sexe, la race, lâge ou la présence dune insuffisance rénale ou hépatique. Il a été observé que lanalyse de la population, qui comprenait des patients âgés de 8 à 72 ans, na eu aucune influence sur les paramètres PK de l'inhibiteur de la C1 estérase.
5.3. Données de sécurité préclinique
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration unique et répétée, tolérance locale et thrombogénicité nont pas révélé de risque particulier pour lHomme après administration intraveineuse ou sous-cutanée.
Aucune étude destinée à évaluer la cancérogénicité et la toxicité reproductrice n'a été réalisée.
6. DONNEES PHARMACEUTIQUES
6.1. Liste des excipients
Poudre :
Glycine, chlorure de sodium, citrate de sodium
Solvant :
Eau pour préparations injectables
6.2. Incompatibilités
En labsence détudes de compatibilité, ce médicament ne doit pas être mélangé avec dautres médicaments.
6.3. Durée de conservation
36 mois
Après reconstitution, la stabilité physico-chimique a été démontrée pendant 48 heures à température ambiante (max. + 30°C). Dun point de vue microbiologique et dans la mesure où BERINERT ne contient aucun conservateur, le produit reconstitué doit être utilisé immédiatement. Si le produit nest pas administré immédiatement, la durée de conservation ne doit pas dépasser 8 heures à température ambiante. Le produit reconstitué doit être conservé uniquement dans le flacon dorigine.
6.4. Précautions particulières de conservation
À conserver à une température ne dépassant pas 30 °C.
Ne pas congeler.
Conserver le flacon dans l'emballage extérieur à l'abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Conditionnements primaires
Poudre (2000 UI) dans un flacon (verre de type II) avec un bouchon (caoutchouc de bromobutyle), un opercule en aluminium et un capuchon amovible en plastique.
4 ml de solvant dans un flacon (verre de type I) avec un bouchon (caoutchouc de chlorobutyle), un opercule en aluminium et un capuchon amovible en plastique.
Présentations :
Une boîte contient :
1 flacon de poudre
1 flacon de solvant (BERINERT 2000 UI : 4 ml)
1 dispositif de transfert de filtre 20/20
Un set dadministration (boîte intérieure) contient :
1 seringue à usage unique (BERINERT 2000 : 5 ml)
1 aiguille hypodermique
1 dispositif dinjection sous-cutanée
2 tampons imbibés dalcool,
1 pansement
Emballage multiple contenant 5 flacons de 2000 UI et 20 flacons de 2000 UI.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Mode dadministration
Instructions générales
· La solution reconstituée de BERINERT doit être incolore et limpide ou légèrement opalescente.
· Après filtration/prélèvement (voir ci-dessous), le produit reconstitué doit être inspecté visuellement afin de mettre en évidence la présence de particules ou un changement de coloration avant administration.
· Nutilisez pas de solutions troubles ou contenant des dépôts.
· La reconstitution et le prélèvement doivent être effectués dans des conditions aseptiques. Utilisez la seringue fournie avec le produit.
Reconstitution
Amenez le solvant à température ambiante. Pensez à retirer les capuchons protecteurs des flacons de produit et de solvant et à nettoyer les bouchons avec une solution antiseptique puis à les laisser sécher avant de procéder à louverture de lemballage du dispositif Mix2vial.
|
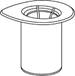 1 1
|
1. Ouvrez lemballage du dispositif Mix2Vial en retirant lopercule. Nenlevez pas le dispositif Mix2Vial de son emballage!
|
|
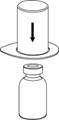 2 2
|
2. Placez le flacon de solvant sur une surface plane et propre et maintenez-le fermement. Saisissez le dispositif Mix2Vial avec son emballage thermoformé et enfoncez lextrémité bleue de ladaptateur tout droit à travers le bouchon du flacon de solvant.
|
|
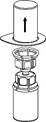 3 3
|
3. Retirez délicatement lemballage thermoformé du dispositif Mix2Vial en le tenant par les bords et en tirant verticalement vers le haut. Veiller à retirer uniquement lemballage thermoformé en laissant le dispositif Mix2Vial en place.
|
|
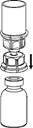 4 4
|
4. Posez le flacon de produit sur une surface plane et rigide. Retournez le flacon de solvant muni du dispositif Mix2Vial et enfoncez lextrémité transparente de ladaptateur tout droit à travers le bouchon du flacon de produit. Le solvant coule automatiquement dans le flacon de produit.
|
|
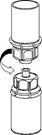 5 5
|
5. En maintenant la partie produit du dispositif Mix2Vial dune main et la partie solvant de lautre, dévissez soigneusement le dispositif dans le sens inverse des aiguilles dune montre afin de séparer les deux flacons.
Jetez le flacon de solvant muni de la partie bleue de ladaptateur Mix2Vial.
|
|
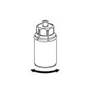 6 6
|
6. Agitez délicatement le flacon de produit muni de la partie transparente de ladaptateur jusquà ce que la substance soit totalement dissoute. Ne secouez pas le flacon.
|
|
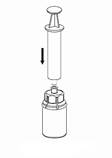 7 7
|
7. Remplissez dair une seringue stérile vide. Utilisez la seringue fournie avec le produit. Tout en maintenant verticalement le flacon de produit, connectez la seringue au raccord Luer Lock du dispositif Mix2Vial en le vissant dans le sens des aiguilles d'une montre. Injectez de lair dans le flacon de produit.
|
Prélèvement et administration
|
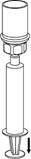 8 8
|
8. Tout en maintenant le piston de la seringue enfoncé, retournez le système et prélevez la solution dans la seringue en tirant lentement sur le piston.
|
|
 9 9
|
9. Une fois la solution transférée dans la seringue, tenez fermement le corps de la seringue (en maintenant le piston de la seringue dirigé vers le bas) et déconnectez la partie transparente de ladaptateur Mix2Vial de la seringue en dévissant dans le sens inverse des aiguilles dune montre.
|
Administration
Le produit peut être administré à laide d'une aiguille hypodermique ou dun kit de perfusion sous-cutanée.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
CSL Behring GmbH
Emil-von-Behring-Strasse 76
35041 Marburg
ALLEMAGNE
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 550 537 1 2 : 2000 UI de poudre en flacon (verre de type II) muni dun bouchon (bromobutyle) + 4 ml de solvant en flacon (verre de type I) muni dun bouchon (chlorobutyle) + un set dadministration. Boîte de 1.
· 34009 550 537 2 9 : 2000 UI de poudre en flacon (verre de type II) muni dun bouchon (bromobutyle) + 4 ml de solvant en flacon (verre de type I) muni dun bouchon (chlorobutyle) + un set dadministration. Boîte de 5.
· 34009 550 537 3 6 : 2000 UI de poudre en flacon (verre de type II) muni dun bouchon (bromobutyle) + 4 ml de solvant en flacon (verre de type I) muni dun bouchon (chlorobutyle) + un set dadministration. Boîte de 20.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
11. DOSIMETRIE
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I
Médicament soumis à prescription hospitalière.