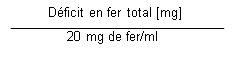RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 30/10/2020
![]() Ce médicament fait lobjet dune surveillance supplémentaire qui permettra lidentification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait lobjet dune surveillance supplémentaire qui permettra lidentification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
FER BAXTER 20 mg/mL, solution pour injection/perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Complexe dhydroxyde ferrique-saccharose..................................................................... 333,00 mg
Quantité correspondant à fer élément. ............................................................................... 20,00 mg
Pour 1 ml de solution injectable.
Une ampoule de 5 ml de FER BAXTER 20 mg/mL, solution pour injection/perfusion contient 1665 mg de complexe dhydroxyde ferrique-saccharose correspondant à 100 mg de fer élément.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution pour injection/perfusion.
FER BAXTER est une solution opaque de couleur brun foncé légèrement visqueuse dont le pH se trouve entre 10,5 et 11,0 à 20°C et dont losmolalité est de 1 150 à 1 350 mOsmol/kg.
4.1. Indications thérapeutiques
FER BAXTER est indiqué dans le traitement, par voie intraveineuse, de l'anémie par carence martiale dans les cas suivants :
· Lorsquil y a un besoin clinique qui nécessite un apport rapide en fer.
· Chez les patients intolérants au fer administré par voie orale ou qui sont non-observants.
· Maladies gastro-intestinale inflammatoires pour lesquelles les traitements oraux à base de fer sont inefficaces.
· Chez linsuffisant rénal chronique lorsquun traitement par fer oral sest révélé insuffisant ou mal toléré.
FER BAXTER ne doit être administré que dans le cas où l'indication est confirmée par des investigations adéquates. Les analyses de laboratoire appropriées sont le dosage de l'hémoglobine, de la ferritine sérique et de la saturation de la transferrine.
4.2. Posologie et mode d'administration
Surveiller attentivement les patients afin de détecter tout signe et symptôme de réactions d'hypersensibilité pendant et après chaque administration de FER BAXTER.
Posologie
La dose cumulée de FER BAXTER doit être calculée individuellement pour chaque patient et ne doit pas être dépassée.
Calcul de la dose totale nécessaire
La dose cumulée totale de FER BAXTER, équivalente au déficit en fer total (mg), est déterminée par le taux dhémoglobine (Hb) et le poids corporel (PC). La dose de FER BAXTER doit être calculée individuellement pour chaque patient en fonction du déficit en fer total calculé selon la formule de Ganzoni ci-dessous, par exemple :
Déficit en fer total [mg] = PC [kg] x (Hb cible Hb actuelle) [g/dl] x 2,4* + réserves de fer [mg]
PC < 35 kg : Hb cible = 13 g/dl et réserves de fer = 15 mg/kg de PC
PC ≥ 35 kg : Hb cible = 15 g/dl et réserves de fer = 500 mg
* Factor 2,4 = 0,0034 (teneur en fer de lHb = 0,34 %) x 0,07 (volume sanguin = 7 % de PC) x 1 000 (facteur de conversion de [g] en [mg]) x 10
|
Dose totale de FER BAXTER à administrer (en ml) = |
|
|
Quantité totale de FER BAXTER (ml) à administrer en fonction du poids corporel, des taux dhémoglobine actuels et des taux cibles* :
|
Poids corporel |
Quantité totale de FER BAXTER |
|||
|
Hb 6,0 g/dl |
Hb 7,5 g/dl |
Hb 9,0 g/dl |
Hb 10,5 g/dl |
|
|
30 kg |
47,5 ml |
42,5 ml |
37,5 ml |
32,5 ml |
|
35 kg |
62,5 ml |
57,5 ml |
50,0 ml |
45,0 ml |
|
40 kg |
67,5 ml |
60,0 ml |
55,0 ml |
47,5 ml |
|
45 kg |
75,0 ml |
65,0 ml |
57,5 ml |
50,0 ml |
|
50 kg |
80,0 ml |
70,0 ml |
60,0 ml |
52,5 ml |
|
55 kg |
85,0 ml |
75,0 ml |
65,0 ml |
55,0 ml |
|
60 kg |
90,0 ml |
80,0 ml |
67,5 ml |
57,5 ml |
|
65 kg |
95,0 ml |
82,5 ml |
72,5 ml |
60,0 ml |
|
70 kg |
100,0 ml |
87,5 ml |
75,0 ml |
62,5 ml |
|
75 kg |
105,0 ml |
92,5 ml |
80,0 ml |
65,0 ml |
|
80 kg |
112,5 ml |
97,5 ml |
82,5 ml |
67,5 ml |
|
85 kg |
117,5 ml |
102,5 ml |
85,0 ml |
70,0 ml |
|
90 kg |
122,5 ml |
107,5 ml |
90,0 ml |
72,5 ml |
* PC < 35 kg : Hb cible = 13 g/dl
* PC ≥ 35 kg : Hb cible = 15 g/dl
Pour convertir le taux d'Hb (mM) en taux d'Hb (g/dl), multipliez le premier par 1,6.
Si la dose totale requise est supérieure à la dose unique maximale autorisée de 200 mg (par injection) ou 500 mg (par perfusion), la dose totale doit être administrée de manière fractionnée.
Adultes
La posologie est de 5 à 10 mL de FER BAXTER (100 à 200 mg de fer) une à trois fois par semaine. Pour la durée d'administration et le rapport de dilution, voir « Mode dadministration ».
Population pédiatrique
La sécurité et lefficacité de FER BAXTER chez les enfants nont pas été établies.
Par conséquent FER BAXTER nest pas recommandée chez les enfants et adolescents âgés de moins de 18 ans.
Insuffisant rénal chronique
Aucun ajustement de la dose nest nécessaire
Insuffisant hépatique
Voir rubrique 4.4
Mode dadministration
FER BAXTER ne doit être administré que par voie intraveineuse. Il peut être administré en perfusion goutte-à-goutte, en injection lente ou directement dans la ligne veineuse d'un dialyseur.
Perfusion intraveineuse goutte à goutte
FER BAXTER ne doit être dilué que dans une solution de chlorure de sodium (NaCl) stérile à 0,9% m/V. La dilution doit être effectuée juste avant la perfusion et la solution doit être administrée comme suit :
|
Dose de FER BAXTER |
Dose de FER BAXTER |
Volume de dilution maximal de la solution de NaCl stérile à 0,9% m/V |
Durée minimale de perfusion |
|
50 mg |
2,5 ml |
50 ml |
8 minutes |
|
100 mg |
5 ml |
100 ml |
15 minutes |
|
200 mg |
10 ml |
200 ml |
30 minutes |
Pour des raisons de stabilité, les dilutions à des concentrations inférieures de FER BAXTER ne sont pas admissibles.
Injection intraveineuse
FER BAXTER peut être administré en injection intraveineuse lente à la vitesse de 1 ml de solution non diluée par minute, sans dépasser 10 ml (200 mg de fer) par injection.
Injection dans la ligne veineuse d'un dialyseur
FER BAXTER peut être administré directement dans la ligne veineuse d'un dialyseur au cours d'une séance d'hémodialyse, dans les mêmes conditions que celles d'une injection intraveineuse.
Lutilisation de FER BAXTER est contre-indiquée dans les situations suivantes :
· hypersensibilité à la substance active ou à lun des excipients mentionnés à la rubrique 6.1,
· hypersensibilité grave connue à toute autre préparation parentérale contenant du fer,
· anémie non ferriprive,
· surcharge martiale ou troubles héréditaires de l'utilisation du fer.
4.4. Mises en garde spéciales et précautions d'emploi
Les préparations à base de fer administrées par voie parentérale sont susceptibles de provoquer des réactions dhypersensibilité, y compris des réactions anaphylactiques/anaphylactoïdes graves et potentiellement mortelles.
Des réactions dhypersensibilité ont également été rapportées après ladministration de doses complexes de fer par voie parentérale, y compris de complexe dhydroxyde ferrique-saccharose, qui navaient précédemment provoqué aucune réaction. Des réactions dhypersensibilité ayant progressé vers un syndrome de Kounis (artériospasme coronaire allergique aigu qui peut donner lieu à un infarctus du myocarde, voir rubrique 4.8) ont été rapportées. Cependant, dans plusieurs études cliniques réalisées chez des patients qui avaient des antécédents de réactions dhypersensibilité au fer dextran ou au gluconate ferrique, le FER BAXTER a montré quil était bien toléré. Pour les cas graves connus dhypersensibilité aux autres préparations parentérales à base de fer, voir rubrique 4.3.
Ce risque de réactions dhypersensibilité est plus élevé chez les patients présentant des allergies connues, y compris des allergies médicamenteuses, des antécédents dasthme, deczéma ou de tout autre type dallergie (terrain atopique) sévères.
Le risque de réactions d'hypersensibilité aux complexes de fers administrés par voie parentérale est également accru chez les patients atteints de troubles immunitaires ou inflammatoires (p. ex. lupus érythémateux systémique, polyarthrite rhumatoïde).
FER BAXTER ne doit être administré que lorsque du personnel formé pour évaluer et prendre en charge les réactions anaphylactiques est immédiatement disponible, dans un environnement disposant des moyens nécessaires pour assurer une réanimation. Le patient doit être surveillé afin de détecter l'apparition de tout effet indésirable pendant au moins 30 minutes après chaque administration de FER BAXTER. Si des manifestations dhypersensibilité ou dintolérance sont observées durant ladministration, le traitement doit être immédiatement arrêté. La prise en charge dune réaction anaphylactique/anaphylactoïde implique davoir à disposition les moyens nécessaires à une réanimation cardio-respiratoire incluant ladrénaline injectable (1 :1 000). Un traitement additionnel par antihistaminique et/ou corticostéroïde peut également savérer nécessaire.
Chez les patients présentant une insuffisance hépatique, le fer parentéral ne doit être administré quaprès une évaluation attentive du rapport bénéfice/risque. Ladministration de fer par voie parentérale doit être évitée chez les patients présentant une insuffisance hépatique lorsque la surcharge en fer constitue un facteur précipitant, en particulier Porphyrie Cutanée Tardive. Une surveillance étroite du bilan martial est recommandée afin déviter une surcharge en fer.
Les préparations à base de fer administrées par voie parentérale doivent être utilisées avec précaution en cas dinfection aiguë ou chronique. Il est recommandé darrêter le traitement par FER BAXTER chez les patients présentant une bactériémie. Le rapport bénéfice/risque doit être évalué chez les patients présentant une infection chronique.
Il convient déviter un écoulement périveineux car lextravasation de FER BAXTER au site dinjection peut provoquer une douleur, une inflammation, une nécrose tissulaire et une pigmentation brune de la peau qui peut persister au point d'injection.
Ce produit contient moins de 1 mmol de sodium (23 mg) par ampoule de 5 ml, cest-à-dire quil est essentiellement sans sodium.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Comme toutes les préparations parentérales à base de fer, FER BAXTER ne doit pas être administré en même temps que des préparations orales de fer, car l'absorption du fer administré par voie orale pourrait être réduite.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'existe pas de données suffisantes sur l'utilisation du fer-saccharose au cours du premier trimestre de grossesse. Des données relatives à l'utilisation de FER BAXTER au cours du deuxième et troisième trimestre de grossesse (303 issues de grossesses) n'ont occasionné aucune préoccupation en matière de sécurité pour la mère ou le nouveau-né.
Une évaluation prudente du rapport bénéfice/risque devra être effectuée avant toute utilisation de FER BAXTER pendant la grossesse ; FER BAXTER ne doit pas être utilisé pendant la grossesse à moins dune nécessité absolue (voir rubrique 4.4).
Une bradycardie ftale peut survenir après ladministration de fers parentéraux. Celle-ci est généralement transitoire et la conséquence dune réaction dhypersensibilité chez la mère. Le bébé à naître doit être étroitement surveillé lors de l'administration par voie intraveineuse de fers parentéraux à la femme enceinte.
Dans bien des cas, lanémie par carence martiale durant le premier trimestre de grossesse peut être traitée par une préparation de fer orale. Lorsque le bénéfice dun traitement par FER BAXTER est estimé supérieur au risque potentiel pour la mère et le ftus, il est recommandé de limiter ce traitement aux deuxième et troisième trimestres.
Les études effectuées chez lanimal nont pas mis en évidence deffets délétères directs ou indirects sur la reproduction (voir rubrique 5.3).
Allaitement
Il existe des données limitées sur l'excrétion du fer dans le lait maternel humain après l'administration de fer-saccharose par voie intraveineuse. Dans une étude clinique, 10 mères qui allaitaient, atteintes de carence en fer et par ailleurs en bonne santé, ont reçu 100 mg de fer sous forme de fer-saccharose.
Quatre jours après le traitement, la teneur en fer du lait maternel n'avait pas augmenté et aucune différence n'était observée par rapport au groupe témoin (n = 5). On ne peut exclure que les nouveau-nés/nourrissons puissent être exposés au fer dérivé de FER BAXTER via le lait maternel. Par conséquent, le rapport bénéfice/risque doit être évalué.
Les données précliniques ne montrent aucun effet nocif direct ou indirect sur l'enfant allaité. Chez les rates en lactation traitées par fer-saccharose marqué au 59Fe, une faible excrétion de fer dans le lait et un faible transfert du fer à la portée ont été observés. Il est peu probable que du fer-saccharose non métabolisé passe dans le lait maternel.
Fertilité
Aucun effet du complexe hydroxyde ferrique-saccharose na été observé sur la fertilité, les performances daccouplement et le développement embryonnaire précoce chez le rat.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
En cas de survenue de symptômes tels que vertiges, confusion ou étourdissements après ladministration de FER BAXTER, les patients ne doivent pas conduire des véhicules ni utiliser des machines jusquà la régression des symptômes.
Leffet indésirable le plus fréquemment rapporté dans les études cliniques menées avec le fer saccharose a été une dysgueusie, survenue avec une fréquence de 4,5 %. Les effets indésirables graves les plus importants associés au fer saccharose sont des réactions dhypersensibilité, qui sont survenues avec une fréquence de 0,25 % dans les études cliniques (voir rubrique 4.4).
Le tableau ci-dessous présente les effets indésirables rapportés après ladministration du fer saccharose chez 4 064 patients dans les études cliniques ainsi que ceux notifiés dans le cadre de la pharmacovigilance post-commercialisation. Les effets indésirables ont été classés en fonction de leur incidence en utilisant la classification suivante : Très fréquent (≥ 1/10) ; fréquent (≥ 1/100 < 1/10) ; peu fréquent (≥ 1/1000 < 1/100) ; rare (≥ 1/10 000 < 1/1000) ; très rare (<1/10 000).
|
Classe de système dorganes |
Fréquent (≥ 1/100, < 1/10) |
Peu fréquent (≥ 1/1000, < 1/100) |
Rare (≥ 1/10 000, < 1/1000) |
Fréquence indéterminée1) |
|
Affections du système immunitaire |
Hypersensibilité |
Angio-dème, réactions anaphylactoïdes |
||
|
Affections du système nerveux |
Dysgueusie |
Céphalées, vertiges, paresthésies, hypoesthésie |
Syncope, somnolence |
Diminution du niveau de conscience, confusion, perte de conscience, anxiété, tremblements |
|
Affections cardiaques |
Palpitations |
Bradycardie, tachycardie, syndrome de Kounis |
||
|
Affections vasculaires |
Hypotension, hypertension |
Bouffées vasomotrices, phlébite |
Collapsus circulatoire, Thrombophlébite |
|
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée |
Bronchospasme |
||
|
Affections du rein et des voies urinaires |
Chromaturie |
|||
|
Affections gastro-intestinales |
Nausées |
Vomissements, douleur abdominale, diarrhée, constipation |
||
|
Affections de la peau et du tissu sous-cutané |
Prurit, rash |
Urticaire, érythème |
||
|
Affections musculosquelettiques et systémiques |
Spasmes musculaires, myalgies, arthralgies, douleurs dans les membres, dorsalgies |
|||
|
Troubles généraux et anomalies au site dadministration |
Réactions au point dinjection/de perfusion2) |
Frissons, asthénie, fatigue, dème périphérique, douleur |
Douleur (ou gêne) thoracique, hyperhidrose, pyrexie |
Syndrome pseudo-grippal apparaissant en quelques heures jusquà quelques jours, Sueurs froides, malaise, pâleur |
|
Investigations |
Augmentation de lalanine amino-transférase, augmentation de laspartate amino-transférase, augmentation de la gamma-glutamyltransférase, augmentation de la ferritine sérique |
Augmentation du taux sanguin de lactate déshydrogénase |
1) Notifications spontanées dans le cadre de la pharmacovigilance.
2) Les effets secondaires les plus fréquents sont les suivants : douleur au point dinjection/de perfusion, extravasation, irritation, réaction, décoloration, hématome, prurit.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Un surdosage peut causer une surcharge martiale pouvant se manifester par une hémosidérose. Si le médecin traitant le juge nécessaire, un surdosage doit être traité en administrant un chélateur du fer ou conformément à la pratique médicale habituelle.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Préparations antianémiques, fer, préparations parentérales, code ATC : B03AC
Mécanisme daction
Le fer-saccharose, la substance active de FER BAXTER, est composé dun noyau dhydroxyde de fer (III) polynucléaire entouré dun grand nombre de molécules de saccharose liées de façon non covalente. Le poids moléculaire (PM) moyen du complexe est denviron 43 kDa. Le noyau de fer polynucléaire possède une structure similaire à celle du noyau de la ferritine, la protéine de stockage du fer physiologique. Le complexe est conçu pour délivrer, de façon contrôlée, le fer utilisable pour les protéines de transport et de stockage du fer dans lorganisme (transferrine et ferritine respectivement).
Après une administration intraveineuse, le noyau de fer polynucléaire du complexe est capté essentiellement par le système réticulo-endothélial dans le foie, la rate et la moelle osseuse. Dans une seconde étape, le fer est utilisé pour la synthèse de lhémoglobine, de la myoglobine et dautres enzymes contenant du fer, ou stocké principalement dans le foie sous forme de ferritine.
Efficacité et sécurité clinique
Insuffisance rénale chronique
Létude LU98001 était une étude à bras unique pour étudier l'efficacité et la tolérance de 100 mg de fer injecté sous forme de saccharose de fer jusqu'à 10 séances sur 3 à 4 semaines chez des patients hémodialysés souffrant d'anémie ferriprive (Hb > 8 et < 11,0 g/dl, TSAT < 20%, et ferritine sérique ≤ 300 μg/l) qui recevaient une thérapie à base dérythropoïétine recombinante humaine. Un taux de Hb ≥ 11 g/dl a été atteint chez 60/77 patients. L'augmentation moyenne de la ferritine sérique et de la TSAT a été significative de la baseline à la fin du traitement (jour 24) ainsi qu'à la visite de suivi de 2 et 5 semaines.
L'étude 1VEN03027 était une étude randomisée comparant l'injection de saccharose de fer (1000 mg en doses fractionnées sur 14 jours) et le sulfate ferreux oral (325 mg 3 fois par jour pendant 56 jours) chez des patients souffrant de maladies rénales chroniques non dialysés (Hb ≤ 11,0 g/dl, ferritine sérique ≤ 300μg/l et TSAT ≤ 25%) avec ou sans érythropoïétine recombinante humaine. Une réponse clinique (définie comme une augmentation de Hb ≥ 1,0 g/dl et une augmentation de la ferritine sérique ≥ 160 μg/l) a été observée plus fréquemment chez les patients traités par injection de saccharose de fer (31/79; 39,2%) comparativement au fer oral (1/82; 1,2%); p < 0,0001.
Maladie gastro-intestinale inflammatoire
Une étude randomisée et contrôlée a comparé l'injection de saccharose de fer (dose intraveineuse unique de 200 mg de fer une fois par semaine ou toutes les deux semaines jusqu'à ce que la dose cumulative ait été atteinte) avec du fer oral (200 mg deux fois par jour pendant 20 semaines) chez des patients souffrant de maladie gastro-intestinale inflammatoire et danémie (Hb < 11,5 g/dl). À la fin du traitement, 66% des patients du groupe d'injection de saccharose de fer avaient une augmentation de Hb ≥ 2,0 g/dl comparativement à 47% dans le groupe de fer oral (p = 0,07).
Postpartum
Une étude randomisée et contrôlée chez des femmes présentant une anémie ferriprive postpartum (Hb < 9 g/dl et ferritine sérique < 15 μg/l à 24 48 heures après laccouchement) a comparé 2 × 200 mg de fer donné sous forme dinjection de saccharose de fer sur les jours 2 et 4 (n = 22) et 200 mg de fer oral donné sous forme de sulphate ferreux deux fois par jour pendant 6 semaines (n = 21). L'augmentation moyenne du taux de Hb de la baseline au jour 5 était de 2,5 g/dl dans le groupe d'injection de saccharose de fer et de 0,7 g/dl dans le groupe de fer oral (p < 0,01).
Grossesse
Dans une étude contrôlée randomisée, des femmes au cours de leur troisième trimestre de grossesse avec une anémie ferriprive (Hb 8 à 10,5 g/dl et ferritine sérique < 13 µg/l) ont été randomisées pour recevoir l'injection de saccharose de fer (dose totale calculée individuellement de fer administré sur 5 jours) ou le complexe de polymaltose de fer oral (100 mg 3 fois par jour jusqu'à laccouchement). L'augmentation du taux de Hb par rapport au niveau de référence était significativement plus élevée dans le groupe d'injection de saccharose de fer que dans le groupe de fer oral au jour 28 et à l'accouchement (p < 0,01).
5.2. Propriétés pharmacocinétiques
Distribution
La ferrocinétique du complexe dhydroxyde ferrique-saccharose marqué au 52Fe et au 59Fe a été évaluée chez 6 patients présentant une insuffisance rénale chronique et une anémie. Au cours des 6 à 8 premières heures, le 52Fe a été capté par le foie, la rate et la moelle osseuse. La captation de la radioactivité par la rate riche en macrophages est considérée comme représentative de la captation du fer par le système réticulo-endothélial.
Après injection intraveineuse dune dose unique de 100 mg de fer sous forme de complexe dhydroxyde ferrique-saccharose chez des volontaires sains, les concentrations sériques maximales de fer totales sont atteintes 10 minutes après linjection, avec une concentration moyenne de 538 µmol/l. Le volume de distribution dans le compartiment central correspond bien au volume plasmatique (environ 3 litres).
Biotransformation
Après linjection, le saccharose se dissocie fortement et le noyau de fer polynucléaire est capté principalement par le système réticulo-endothélial du foie, de la rate et de la moelle osseuse. Quatre semaines après ladministration, lutilisation du fer érythrocytaire est de 59 % à 97 %.
Élimination
Le poids moléculaire (PM) moyen du complexe dhydroxyde ferrique-saccharose est denviron 43 kDa, soit un poids suffisamment important pour empêcher lélimination par voie rénale. Lélimination rénale du fer, qui se produit pendant les 4 premières heures suivant linjection dune dose de 100 mg de fer sous forme de FER BAXTER, représente moins de 5 % de la dose. Après 24 heures, la concentration sérique totale de fer est diminuée à la valeur avant ladministration et lélimination rénale du saccharose représente environ 75 % de la dose administrée.
5.3. Données de sécurité préclinique
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité, cancérogénèse, et des fonctions de reproduction et de développement, nont pas révélé de risque particulier pour lhomme.
Hydroxyde de sodium (pour lajustement du pH), eau pour préparations injectables, azote (gaz inerte).
Ce médicament ne doit pas être mélangé avec dautres médicaments à lexception de ceux mentionnés dans la rubrique 6.6.
Une précipitation et/ou une interaction sont possibles en cas de mélange avec dautres solutions ou médicaments. La compatibilité avec les récipients autres quen verre, en polyéthylène et en PVC nest pas connue.
Durée de conservation du produit tel que conditionné pour la vente
3 ans.
Durée de conservation après première ouverture de lampoule
Dun point de vue microbiologique, le produit doit être utilisé immédiatement.
Durée de conservation après dilution avec une solution stérile de chlorure de sodium (NaCl) à 0,9 % m/v
Dun point de vue microbiologique, le produit doit être utilisé immédiatement après dilution avec une solution stérile de chlorure de sodium (NaCl) à 0,9 % m/v.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas à 25°C.
Ne pas congeler.
A conserver dans lemballage dorigine, à labri de la lumière.
Pour les conditions de conservation du médicament après dilution ou première ouverture, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
5 ml en ampoule (verre de type I). Boîte de 5, 10 ou 25.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Les ampoules doivent être examinées avant lutilisation pour vérifier labsence de sédiments ou de dommage. Nutiliser que les ampoules contenant une solution homogène et exempte de sédiments.
FER BAXTER ne doit pas être mélangé avec dautres médicaments, à lexception dune solution stérile de chlorure de sodium à 0,9 % m/v pour la dilution. Pour des instructions sur la dilution du produit avant ladministration, voir la rubrique 4.2.
La solution diluée doit être de couleur brune et limpide.
Chaque ampoule de FER BAXTER est à usage unique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
KOBALTWEG 49
3542CE UTRECHT
PAYS-BAS
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 550 596 7 7 : Solution pour injection/perfusion en ampoule de 5 mL (verre). Boîte de 5.
· 34009 550 596 8 4 : Solution pour injection/perfusion en ampoule de 5 mL (verre). Boîte de 10.
· 34009 550 596 9 1 : Solution pour injection/perfusion en ampoule de 5 mL (verre). Boîte de 25.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I.
Médicament réservé à lusage hospitalier.