RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 07/06/2024
MIGOUTINE 0,5 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé pelliculé contient 0,5 mg de colchicine.
Excipients à effet notoire :
Chaque comprimé contient:
· 104,62 mg de saccharose
· 2,99 mg de maltodextrine
· 1,81 mg de sirop de sucre inverti
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé de couleur bleu-vert, au fini translucide à blanc, de forme rond, biconvexe (environ 6,4 x 3,3 mm), gravé « P » sur une face et « 05 » sur l'autre face.
4.1. Indications thérapeutiques
Prévention des événements cardiovasculaires ischémiques chez les patients atteints de maladie coronarienne liée à lathérosclérose après un infarctus du myocarde (IM) récent, en appoint aux traitements standards.
4.2. Posologie et mode d'administration
Posologie
Adultes:
La dose recommandée est de 0,5 mg (1 comprimé) une fois par jour. Chez les patients présentant une maladie coronarienne établie, le traitement doit être initié dans les 3 jours suivant linfarctus du myocarde. Il ne doit pas être initié au delà de 30 jours après linfarctus du myocarde. La dose maximale est de 0,5 mg par jour.
Durée du traitement : la sécurité demploi de Migoutine est établie pour une période de 2 ans sur la base de l'étude COLCOT (voir rubrique 5.1).
Population âgée:
Les données issues des études cliniques et de l'expérience suggèrent que l'utilisation chez les patients âgés est associée à des différences en termes de sécurité ou d'efficacité. En raison de l'incidence accrue de l'insuffisance rénale et de l'incidence plus élevée d'autres comorbidités, nécessitant l'utilisation d'autres médicaments, Migoutine doit être utilisé avec prudence chez les personnes âgées.
Population pédiatrique :
Il n'existe aucune utilisation pertinente de Migoutine dans la population pédiatrique pour la prévention des événements cardiovasculaires ischémiques chez les patients présentant une maladie coronarienne liée à lathérosclérose après un infarctus du myocarde récent.
Patients présentant une insuffisance rénale :
Les patients présentant une insuffisance rénale doivent faire l'objet d'une surveillance étroite visant à détecter tout effet indésirable éventuel de la colchicine (voir rubrique 5.2). Quel que soit le degré d'insuffisance rénale, Migoutine ne doit pas être administré en association avec des inhibiteurs puissants de la P-gp ou des inhibiteurs puissants du CYP3A4. Migoutine est contre-indiqué chez les patients présentant une insuffisance rénale sévère (voir rubrique 4.3).
Patients présentant une insuffisance hépatique :
Les patients présentant une insuffisance hépatique doivent faire l'objet d'une surveillance étroite visant à détecter tout effet indésirable éventuel de la colchicine (voir rubrique 5.2). Quel que soit le degré d'insuffisance hépatique, Migoutine ne doit pas être administré en association avec de puissants inhibiteurs de la P-gp ou de puissants inhibiteurs du CYP3A4. Migoutine est contre-indiqué chez les patients présentant une insuffisance hépatique sévère (voir rubrique 4.3).
Mode dadministration
Pour administration orale. Migoutine peut être pris pendant ou en dehors des repas. Les comprimés doivent être avalés entiers avec un verre d'eau.
Si une dose est oubliée, les patients doivent la prendre dès qu'ils s'en rendent compte. Cependant, sils oublient une dose de Migoutine, ils ne doivent pas doubler la dose lors de la prochaine prise.
· Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1.
· Insuffisance rénale sévère (DFGe < 30 ml/min)
· Insuffisance hépatique sévère
· Dyscrasies sanguines existantes
· Administration concomitante avec des inhibiteurs puissants de la glycoprotéine P ou du CYP3A4 (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
La colchicine a une marge thérapeutique étroite, ce qui la rend susceptible de présenter des risques importants de surdosage sévère. La posologie maximale recommandée ne doit pas être dépassée. Les premières manifestations cliniques d'un tel surdosage comprennent des troubles gastro-intestinaux tels que diarrhée, nausées et vomissements. En fonction de la dose ingérée, une défaillance de plusieurs organes peut survenir, affectant divers systèmes, notamment respiratoire, cardiovasculaire, hématologique et neurologique. Les patients doivent être informés de ces signes de surdosage potentiel, qui peuvent également survenir si les interactions médicamenteuses ne sont pas prises en compte. Une réduction de la posologie ou un arrêt du traitement doit alors être envisagé. Veuillez-vous référer aux rubriques 4.4, 4.5., 4.8 et 4.9 de ce RCP.
Le médicament doit être conservé hors de portée des tiers avant et après utilisation.
Système cardiovasculaire
Il n'existe aucune donnée concernant l'efficacité et la sécurité demploi de la colchicine chez les patients atteints d'insuffisance cardiaque de classe III-IV de la New York Heart Association et présentant une fraction d'éjection ventriculaire gauche inférieure à 35 % (NYHA III-IV, FEVG < 35 %). Voir rubrique 5.1 pour toutes les populations exclues de l'étude COLCOT, pour lesquelles les données cliniques sur l'efficacité et la sécurité demploi ne sont pas disponibles.
Appareil digestif
Les troubles gastro-intestinaux sont les effets indésirables les plus fréquents de la colchicine. Il sagit souvent des premiers signes dune toxicité pouvant indiquer que le traitement doit être interrompu. Ces effets indésirables comprennent la diarrhée, les nausées, les vomissements et les douleurs ou les crampes abdominales.
Les patients présentant des maladies gastro-intestinales sous-jacentes importantes, par exemple des maladies inflammatoires de l'intestin, une diarrhée chronique, etc. ne doivent pas être traités par MIGOUTINE.
Système endocrinien et métabolisme
Ladministration concomitante de Migoutine et dun inhibiteur puissant de la P-gp et/ou dun inhibiteur puissant du CYP3A4 augmente l'exposition à la colchicine, ce qui peut entraîner des effets toxiques, potentiellement mortels de ce médicament. Uneutilisation concomitante avec ces médicaments est contre-indiquée (voir rubrique 4.3).
Migoutine doit être utilisé avec prudence chez les patients présentant d'autres facteurs de risque d'exposition systémique accrue à la colchicine, tels qu'une insuffisance rénale ou hépatique modérée , ou chez les patients âgés. Chez ces patients, l'utilisation concomitante de Migoutine et dinhibiteurs modérés du CYP3A4 doit être évitée (voir rubrique 4.5).
Il a été démontré que la colchicine induit une malabsorption réversible de la vitamine B12 (voir rubrique 4.5).
Système sanguin
Des cas de dépression médullaire, de leucopénie, de granulocytopénie, de thrombocytopénie, de pancytopénie et danémie aplasique ont été signalés chez des patients prenant de la colchicine. Il est recommandé deffectuer des analyses de sang régulières, car l'administration prolongée de colchicine peut provoquer des dyscrasies sanguines.
Migoutine est contre-indiqué chez les patients présentant des dyscrasies sanguines préexistantes (voir rubrique 4.3).
Appareil locomoteur
Des cas de toxicité neuromusculaire et de rhabdomyolyse induites par la colchicine ont été rapportés lors de lutilisation prolongée de ce médicament dans d'autres indications (par exemple, goutte chronique). Les patients atteints dinsuffisance rénale et les patients âgés, même ceux ayant des fonctions rénale et hépatique normales, sont exposés à un risque accru de tels effets. L'utilisation concomitante de statines, dont l'atorvastatine, la rosuvastatine et la simvastatine, ainsi que de gemfibrozil, de fénofibrate, d'acide fénofibrique ou de bézafibrate (tous associés à une myotoxicité), ou de cyclosporine, avec la colchicine peut potentialiser le risque de myopathie (voir rubrique 4.5). Après larrêt du traitement par la colchicine, les symptômes disparaissent généralement dans un délai de une semaine à plusieurs mois.
Fonction hépatique
Les patients atteints dinsuffisance hépatique doivent faire l'objet d'une surveillance étroite visant à détecter tout effet indésirable éventuel de la colchicine.
On sait que la colchicine est métabolisée par le foie chez l'homme et la présence d'une insuffisance hépatique sévère a été associée à une toxicité de la colchicine. La clairance hépatique de la colchicine peut être significativement réduite et la demi-vie plasmatique prolongée chez les patients présentant une insuffisance hépatique chronique (voir rubrique 5.2). L'utilisation de Migoutine est contre-indiquée chez les patients présentant une insuffisance hépatique sévère (voir rubrique 4.3).
Migoutine ne doit pas être prescrit en association avec des inhibiteurs puissants de la P-gp ou du CYP3A4, notamment chez les patients présentant une insuffisance hépatique (voir rubriques 4.3 et 4.5). , Des effets toxiques parfois mortels ont été signalés chez de tels patients prenant de la colchicine à des doses thérapeutiques. L'utilisation concomitante d'inhibiteurs modérés du CYP3A4 avec Migoutine doit être évitée chez les patients présentant des facteurs de risque d'exposition systémique accrue à la colchicine, comme une insuffisance hépatique modérée.
Fonction rénale
Les patients atteints dinsuffisance rénale doivent faire l'objet d'une surveillance étroite visant à détecter tout effet indésirable éventuel de la colchicine. On sait que la colchicine est excrétée par voie rénale et la présence d'une insuffisance rénale sévère a été associée à des effets toxiques de la colchicine. Il se peut que lexcrétion urinaire de la colchicine et de ses métabolites soit diminuée en cas dinsuffisance rénale (voir rubrique 5.2).
Les patients présentant une insuffisance rénale légère ou modérée doivent faire lobjet dune surveillance régulière de la fonction rénale et de prudence. Migoutine est contre-indiqué chez les patients atteints dinsuffisance rénale sévère (voir rubrique 4.3).
Migoutine ne doit pas être prescrit en association avec des inhibiteurs puissants de la P-gp ou du CYP3A4, notamment chez les patients atteints dinsuffisance rénale (voir rubriques 4.3 et 4.5). L'utilisation concomitante d'inhibiteurs modérés du CYP3A4 et de Migoutine doit être évitée chez les patients présentant des facteurs de risque augmentant l'exposition systémique à la colchicine, comme une insuffisance rénale modérée.
Hypersensibilité
Ce produit contient du saccharose, de la maltodextrine (glucose) et du sucre inverti. Les patients présentant des problèmes héréditaires rares d'intolérance au fructose, de malabsorption du glucose-galactose ou d'insuffisance en sucrase-isomaltase ne doivent pas prendre ce médicament.
Femmes en âge de procréer / Contraception chez les hommes et les femmes
Compte tenu du mécanisme d'action de la colchicine (voir rubrique 5.3), les femmes en âge de procréer doivent utiliser une méthode de contraception très efficace pendant le traitement par Migoutine et jusqu'à un mois après la fin du traitement. Les patients de sexe masculin ne doivent pas avoir d'enfant pendant le traitement par Migoutine et jusqu'à 3 mois après la dernière dose de traitement.
Autres précautions
Les données des essais cliniques ont montré une tendance à un risque accru de décès non cardiovasculaire chez les patients traités par colchicine. Bien qu'une association claire entre le traitement par la colchicine et les décès non cardiovasculaires n'ait pas été établie, des précautions doivent être prises chez les patients atteints d'une maladie coronarienne chronique traités par la colchicine et présentant des comorbidités pouvant être à l'origine de causes potentielles de mortalité. Il est important de peser les bénéfices et les risques potentiels et de surveiller attentivement les patients afin de détecter tout signe ou symptôme de toxicité.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Interactions médicamenteuses graves
Ladministration concomitante de Migoutine et dun inhibiteur puissant de la P-gp et/ou dun inhibiteur puissant du CYP3A4 augmente l'exposition à la colchicine, ce qui peut entraîner des effets toxiques, potentiellement mortels de ce médicament. Uneutilisation concomitante avec ces médicaments est contre-indiquée (voir rubrique 4.3 et rubrique 4.5, Tableau 1).
Migoutine n'a pas été étudié spécifiquement pour les interactions médicamenteuses, mais les données et informations publiées concernant les interactions médicamenteuses possibles avec la colchicine sont présentées.
La colchicine est un substrat de la glycoprotéine P (P-gp), transporteur d'efflux. Parmi toutes les enzymes du cytochrome P450 évaluées, le CYP3A4 est celui qui intervient principalement dans le métabolisme de la colchicine. Ladministration concomitante de Migoutine et de médicament qui inhibent la P-gp, dont la plupart inhibent également le CYP3A4, entrainera probablement une augmentation des concentrations de colchicine.
Les médecins doivent s'assurer que les patients sont des candidats appropriés pour le traitement par Migoutine et rester attentifs aux signes et symptômes de toxicité liés à une exposition accrue à la colchicine suite à une interaction médicamenteuse. Les signes et symptômes de toxicité de la colchicine doivent être évalués rapidement et, si une toxicité est suspectée, envisager l'interruption ou l'arrêt du traitement par la colchicine.
Les médicaments répertoriés dans le tableau 1 ci-dessous sont basés sur des rapports de cas ou des études dinteractions médicamenteuses, ou sur des interactions potentielles dont on sattend quelles soient intenses et graves (c'est-à-dire celles identifiées comme contre-indiquées).
Table 1 Interactions médicamenteuses établies ou potentielles
|
Classe de médicament ou aliment concomitant |
Résultat noté et attendu |
Commentaire clinique |
|
Inhibiteurs puissants du CYP3A4 : Atazanavir, clarithromycine, darunavir/ritonavir, indinavir, itraconazole, kétoconazole, lopinavir/ritonavir, néfazodone, nelfinavir, ritonavir, saquinavir, télithromycine, tipranavir/ritonavir |
Des hausses significatives de la concentration plasmatique de colchicine ont été observées. Des effetstoxiques mortels de la colchicine ont été signalés lors de lutilisation concomitante avec la clarithromycine, un puissant inhibiteur du CYP3A4. De même, une hausse significative de la concentrationplasmatique de colchicine est à prévoir avec d'autres inhibiteurs puissants du CYP3A4. |
L'utilisation concomitante de Migoutine et dinhibiteurs puissants du CYP3A4 est contre-indiquée (voir rubrique 4.3). |
|
Inhibiteurs modérés du CYP3A4 : Amprénavir, aprépitant, diltiazem, érythromycine, fluconazole, fosamprénavir (promédicament de l'amprénavir), jus de pamplemousse, vérapamil |
Des hausses significatives de la concentration plasmatique de colchicine ont été observées. Des cas detoxicité neuromusculaire ont été signalés à la suite dinteractions avec le diltiazem et le vérapamil. |
Peser les bénéfices et les risques potentiels et surveiller étroitement les patients concernés afin de détecter tout signe ou symptôme de toxicité.
Éviter l'utilisation concomitante de Migoutine avec des inhibiteurs modérés du CYP3A4 chez les patients âgés et chez les patients atteints dinsuffisance rénale ou hépatique modérée. |
|
Inhibiteurs puissants de la P-gp : Cyclosporine, ranolazine |
Des hausses significatives de la concentration plasmatique de colchicine ont été observées. Des effets toxiques mortels de la colchicine ont été signalés lors de lutilisation concomitante avec la cyclosporine, un inhibiteur de la P-gP. De même, une hausse significative de la concentration plasmatique de colchicine est à prévoir avec d'autres inhibiteurs de la P-gp. |
L'utilisation concomitante de Migoutine avec des inhibiteurs puissants de la P-gp est contre-indiquée (voir rubrique 4.3). |
|
Inhibiteurs de lHMG-CoA- réductase : Atorvastatine, rosuvastatine, simvastatine |
Interactions pharmacocinétiques et/ou pharmacodynamiques : l'ajout de la colchicine au traitement stable de longue durée a entraîné une myopathie et une rhabdomyolyse (qui a été parfois mortelle) |
Surveiller étroitement les patients concernés afin de déceler tout signe ou symptôme musculaire, tel quune douleur, une sensibilité ou une faiblesse, plus particulièrement au début du traitement. La surveillance du taux de CPK (créatine phosphokinase) ne préviendra pas nécessairement la survenue dune myopathie sévère. |
|
Autres hypolipidémiants : Fibrates, gemfibrozil |
||
|
Glycosides digitaliques : Digoxine
|
Substrat P-gp ; des cas de rhabdomyolyse ont été signalés |
Interactions médicament-aliment
Lorsque Migoutine a été administré avec de la nourriture dans une étude de biodisponibilité, les résultats ont montré une absence d'effet alimentaire significatif (voir rubrique 5.2). Migoutine peut être administré pendant ou en dehors des repas.
Les données d'une étude sur l'interaction potentielle entre le jus de pamplemousse (un inhibiteur modéré du CYP3A4) et la colchicine suggèrent que le jus de pamplemousse peut augmenter la biodisponibilité orale de la colchicine. Évitez le pamplemousse ou le jus de pamplemousse lorsque vous prenez Migoutine.
Interactions médicament-tests de laboratoire
Il a été démontré que le traitement par la colchicine influe sur les résultats des tests de laboratoire. Les effets qui pourraient avoir une importance clinique sont notamment lobtention de faux positifs à la numération des globules rouges (RBC) et au dosage de l'hémoglobine lors des analyses d'urine à visée diagnostique ainsi que des interactions avec le dosage des 17-hydroxycorticostéroïdes dans l'urine selon la méthode de Reddy, Jenkins et Thorn.
Interaction avec la vitamine B12
L'administration prolongée de colchicine provoque une malabsorption réversible de la vitamine B12.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer / Contraception chez les hommes et les femmes
Compte tenu du mécanisme d'action de la colchicine (voir rubrique 5.3), les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par Migoutine et pendant au moins un mois après l'arrêt du traitement par Migoutine (voir rubrique 4.4). Les patients de sexe masculin ne doivent pas avoir d'enfant pendant le traitement par Migoutine et pendant au moins 3 mois après l'arrêt du traitement par Migoutine (voir rubrique 4.4).
Grossesse
Il n'existe aucune donnée sur l'utilisation de la colchicine chez les femmes enceintes atteintes d'une maladie cardiovasculaire ischémique. Il est à noter que les femmes atteintes dune maladie coronarienne athéroscléreuse préexistante courent un risque accru de complications pendant la grossesse.
Un nombre modéré de données sur les grossesses de femmes traitées par colchicine pour d'autres affections (principalement la fièvre méditerranéenne familiale, FMF) n'ont pas montré d'augmentation du risque de fausse couche ou d'avortement. Cependant, en raison du mécanisme d'action de la colchicine et de rares cas isolés de trisomie 21, la possibilité d'anomalies chromosomiques chez les enfants de parents traités au moment de la conception a été évoquée mais n'est pas confirmée par des études plus récentes. Des études animales ont montré une toxicité pour la reproduction (voir 5.3). La colchicine traverse le placenta humain.
Migoutine ne doit pas être utilisé pendant la grossesse, sauf si l'état clinique de la femme nécessite un traitement par colchicine.
Allaitement
La quantité de colchicine ingérée via le lait est importante : le nourrisson allaité reçoit jusqu'à 10 % de la dose maternelle. Les données de la littérature ne montrent aucun effet indésirable sur un total d'environ 150 enfants exposés pendant l'allaitement. Compte tenu du mécanisme d'action de la colchicine et de son passage dans le lait maternel, un risque pour un nourrisson allaité ne peut être exclu.
Par mesure de précaution, une décision doit être prise entre l'arrêt de l'allaitement ou l'arrêt/l'abstention du traitement par Migoutine en tenant compte du bénéfice de l'allaitement pour l'enfant et du bénéfice du traitement pour la femme.
Fertilité
Des études sur lanimal réalisées avec de la colchicine ont montré des effets indésirables sur la fertilité (voir rubrique 5.3). Des cas rapportés et des études épidémiologiques chez des sujets humains de sexe masculin traités par la colchicine suggèrent que l'infertilité due à la colchicine (telle que l'azoospermie) est rare et peut être réversible. Cependant, la relation causale na pas été clairement établie.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Migoutine na aucune influence sur laptitude à conduire des véhicules et à utiliser des machines.
Les troubles gastro-intestinaux sont les effets indésirables les plus courants liés à la colchicine. Il sagit souvent des premiers signes dune toxicité pouvant indiquer que le traitement doit être interrompu. Ces effets indésirables comprennent la diarrhée, les nausées, les vomissements et les douleurs ou les crampes abdominales.
Effets indésirables post-commercialisation
Les données post-commercialisation sur Migoutine sont limitées.
Des cas de toxicité neuromusculaire induite par la colchicine et pouvant se présenter sous la forme dune douleur ou dune faiblesse musculaires ont été signalés dans le passé (voir rubrique 4.4). Les manifestations toxiques graves suivantes ont été associées à la colchicine : dépression médullaire, coagulation intravasculaire disséminée et atteinte de la fonction rénale, hépatique, de lappareil circulatoire et du système nerveux central. Dans la plupart des cas, ces manifestations toxiques étaient attribuables à une accumulation excessive ou à un surdosage de colchicine (voir rubrique 4.9).
Les effets indésirables suivants, classés par classe d'organes et fréquence, ont été rapportés lors du traitement par la colchicine à long terme dans d'autres indications.
Les fréquences sont définies comme très fréquent (≥ 1/10), fréquent (≥ 1/100 et < 1/10), peu fréquent (≥ 1/1 000 et < 1/100), rare (≥ 1/10 000 et < 1/1 000) et très rare (< 1/10 000) y compris cas isolés. En général, ils étaient réversibles après linterruption temporaire du traitement ou la diminution de la dose de colchicine. Étant donné que ces effets indésirables ont été déclarés spontanément au sein dune population dont la taille est incertaine, il n'est pas possible d'estimer leur fréquence de manière fiable, ni d'établir un lien de causalité entre eux et l'exposition au médicament.
Effets dermatologiques :
Rare : alopécie, éruption maculo-papuleuse, purpura, éruption cutanée
Effets gastro-intestinaux :
Fréquent : crampes abdominales, intolérance au lactose, vomissements
Effets hématologiques :
Peu fréquent : leucopénie, granulocytopénie, thrombocytopénie, pancytopénie, anémie aplasique
Effets hépatobiliaires :
Fréquence indéterminée : AST élevée, ALT élevée, hépatotoxicité
Effets musculosquelettiques :
Peu fréquent : myopathie, CPK élevée, myotonie, faiblesse musculaire, douleurs musculaires ;
Très rare : rhabdomyolyse
Effets neurologiques :
Fréquence indéterminée : neuropathie, névrite périphérique
Effets sur la reproduction :
Fréquence indéterminée : azoospermie, oligospermie
Des réactions indésirables cutanées graves, à savoir le syndrome de Stevens-Johnson (SJS), la nécrolyse épidermique toxique (NET) et le syndrome dhypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), ont été observées au cours du traitement par la colchicine.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
La colchicine a une marge thérapeutique étroite et elle est hautement toxique en cas de surdosage. Des cas de surdose mortelle (accidentelle ou intentionnelle) ont été rapportés chez des adultes et des enfants ayant ingéré de la colchicine.
Migoutine doit être tenu hors de portée des enfants.
Il existe généralement une période de latence de 2 à 12 heures entre le surdosage et l'apparition des symptômes, quelle que soit la voie d'administration. Des décès ont été signalés à une dose aussi faible que 7 mg, bien que des doses plus élevées aient été prises sans sans pour autant savérer létales.
On ignore à partir de quelle dose la colchicine entraîne des effets toxiques importants. Tous les patients qui ont ingéré une surdose de colchicine doivent être évalués immédiatement par un médecin, même en l'absence de symptômes précoces.
Symptômes
La première étape dune intoxication aiguë à la colchicine commence généralement au cours des 24 heures qui suivent l'ingestion et se caractérise par des symptômes gastro-intestinaux tels quune douleur abdominale, des nausées, des vomissements, une diarrhée et une perte de liquide importante, qui entraînent une déplétion volémique. Une leucocytose périphérique peut également être observée.
La deuxième phase, qui est associée à des complications potentiellement mortelles, se déclare de 24 à 72 heures après l'ingestion et se caractérise par les symptômes suivants : défaillance multiviscérale, insuffisance rénale aiguë, confusion, coma, neuropathie périphérique sensitivo-motrice ascendante , dépression myocardique, pancytopénie, dysrythmies, insuffisance respiratoire, coagulopathie de consommation. Le décès survient généralement par suite dune dépression respiratoire et dun collapsus cardiovasculaire. Le rétablissement des patients qui survivent peut être associé à une leucocytose rebond et à une alopécie réversible, lesquelles apparaissent environ une semaine après l'ingestion initiale.
Traitement
Il nexiste aucun antidote spécifique. Il faut arrêter immédiatement le traitement. Pour éliminer les toxines, effectuer un lavage gastrique dans l'heure suivant lintoxication aiguë. Envisager dadministrer, dans lheure qui suit larrivée à lhôpital, du charbon activé par voie orale aux adultes qui ont ingéré plus de 0,1 mg/kg de poids corporel de colchicine, de même quaux enfants qui ont ingéré de la colchicine, peu importe la dose. L'hémodialyse se révèle inefficace dans ce cas (volume de distribution apparent élevé). Une étroite surveillance clinique et biologique étroite en milieu hospitalier simpose.
Traitement symptomatique et traitement de soutien
Ils consistent en la surveillance de la respiration, la stabilisation de la pression artérielle et le maintien de la circulation sanguine, et la correction des déséquilibres hydro-électrolytiques. La dose létale varie considérablement chez ladulte (7 à 65 mg en dose unique), mais elle est généralement d'environ 20 mg.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Remarque : La colchicine indiquée pour la réduction des événements athérothrombotiques est également classée sous le code M04AC01.
Mécanisme d'action
Le mécanisme daction exact de la colchicine dans la prévention secondaire des événements cardiovasculaires majeurs na pas encore été complètement élucidé. Cependant, il a été établit que la colchicine perturbe la fonction cytosquelettique en inhibant la polymérisation des β-tubulines en microtubules et quelle empêche par conséquent l'activation, la dégranulation et la migration des neutrophiles. Des données suggèrent que la colchicine peut également interférer avec la formation intracellulaire des inflammasomes présents dans les neutrophiles et les monocytes (complexes qui sont des médiateurs de l'activation de l'interleukine-1β).
Il a été démontré récemment que la colchicine inhibe l'activation de la caspase-1, composante enzymatique de l'inflammasome de la famille des pyrines 3 (NLRP3) des récepteurs du domaine d'oligomérisation de liaison aux nucléotides (récepteurs de type NOD). La colchicine peut augmenter le seuil d'activation maximale de l'inflammasome, notamment en atténuant (sans pour autant léliminer complètement) l'inflammation subclinique.
Les autres activités anti-inflammatoires potentielles de la colchicine comprennent la modulation de l'expression de la pyrine, la régulation négative de la production d'ARNm du TNF-a induite par les lipopolysaccharides, l'inhibition de la libération d'histamine par les mastocytes, la suppression de la synthèse du procollagène et la promotion de l'activité de la collagénase.
Pharmacodynamie
La pharmacodynamie de la colchicine dans la prévention des événements cardiovasculaires ischémiques n'est pas complètement comprise ; cependant, il a été démontré que la colchicine exerce des effets cardioprotecteurs, anti-inflammatoires et anti-athéroscléreux dans plusieurs modèles animaux in vivo et peut stabiliser les plaques athéroscléreuses en réduisant l'activité inflammatoire et le fardeau athéroscléreux. Des effets favorables (modification des plaques) du traitement quotidien de 0,5 mg de colchicine ont été observés chez des patients ayant présenté un syndrome coronarien aigu, peu importe sil y avait eu ou non une réduction considérable des lipoprotéines de basse densité ou intensification du traitement par une statine à forte dose.
Efficacité et sécurité cliniques
COLCOT (NCT02551094) était un essai international, randomisé, en double aveugle, contrôlé par placebo, multicentrique et axé sur les événements. Au total, 4 745 patients ayant subi un infarctus du myocarde récent ont été recrutés (2 366 patients dans le groupe Migoutine et 2 379 dans le groupe placebo). La durée médiane du traitement par l'étude était de 19,6 mois.
Les patients adultes étaient éligibles pour participer à l'essai COLCOT s'ils avaient subi un infarctus du myocarde (IM) dans les 30 jours précédant l'admission (l'IM « index »), s'ils avaient subi linterventionde revascularisation percutanée prévue (quelle quelle soit) et étaient traités conformément aux recommandations nationales incluant le traitement intensif par une statine. Les patients qui présentaient lune des caractéristiques suivantes n'étaient pas admissibles à létude : maladie mal contrôlée, telle qu'une insuffisance cardiaque de classe III-IV, fraction d'éjection ventriculaire gauche < 35 %, antécédent récent daccident vasculaire cérébral (au cours des 3 derniers mois), IM d'indice de type 2 (secondaire à déséquilibre ischémique), pontage aorto-coronarien planifié ou déjà réalisé (au cours des 3 dernières années), antécédents de cancer ou de maladie lymphoproliférative au cours des 3 dernières années, maladie inflammatoire de l'intestin (maladie de Crohn ou colite ulcéreuse), diarrhée chronique, maladie neuromusculaire évolutive préexistante, anomalies de certains paramètres de laboratoire, prise concomitantes de certains traitements, antécédents de cirrhose, d'hépatite chronique évolutive ou de maladie hépatique grave, grossesse ou allaitaient.
Les caractéristiques démographiques et initiales des patients étaient bien équilibrées et aucune différence cliniquement significative na été observée lors de la comparaison des différents groupes de traitement. La majorité des patients étaient des hommes (80,8 %) et de race blanche (72,5 %). L'âge moyen était de 60,6 ans (ET = 10,7) avec une tranche d'âge comprise entre 20 et 94 ans. LIMC moyen était de 28,3 kg/m2, sans différence observée entre les groupes. Les patients ont été randomisés en moyenne 13,5 jours après l'IM et la plupart d'entre eux (93,0 %) ont subi une intervention coronarienne percutanée (ICP) pour leur IM d'index.
Aucune différence cliniquement significative concernant les antécédents médicaux et les médicaments concomitants n'a été observée lors de la comparaison des différents groupes de traitement. La plupart des patients étaient soit fumeurs (29,9 %), soit avaient déjà fumé (36,7 %), avaient des antécédents d'hypertension (51,0 %) ou de dyslipidémie (45,0 %). La majorité des patients ont reçu des soins médicaux standards comprenant des agents antithrombotiques (99,8 %), des régulateurs du métabolisme des lipides (99,3 %) et des bêtabloquants (88,9 %).
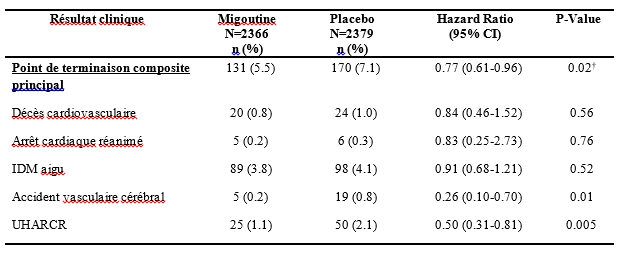
Le critère d'évaluation principal était le temps écoulé entre la randomisation et le premier événement cardiovasculaire tel que le décès dorigine cardiovasculaire, l'arrêt cardiaque avec réanimation, l'IM aigu, l'accident vasculaire cérébral ou l'hospitalisation urgente pour angor nécessitant une revascularisation coronarienne (UHARCR). Le critère d'évaluation principal a été comparé entre les deux groupes de l'essai à l'aide d'un test du log-rank, et le rapport de risque, avec un intervalle de confiance de 95 %, a été calculé à partir d'un modèle à risques proportionnels de Cox.
Les critères d'évaluation secondaires comprenaient le temps écoulé jusqu'à la mortalité totale, les composantes du critère d'évaluation principal et l'ensemble des décès cardiovasculaires, des arrêts cardiaques réanimés, des infarctus du myocarde aigus ou des accidents vasculaires cérébraux. Les événements cardiovasculaires récurrents ont également été lobjet dune évaluation prédéfinie.
Le critère d'évaluation principal est survenu chez 5,5 % des patients traités par Migoutine et 7,1 % du groupe placebo (voir Tableau 3). La différence entre les groupes de traitement était statistiquement significative (HR : 0,77 ; IC à 95 %, 0,61 à 0,96 ; p = 0,02).
Table 2 Résultats COLCOT pour les principaux résultats cliniques (population en intention de traiter)
|
|
Le test du log-rank et le modèle multivarié à risque proportionnel de Cox incluant l'âge, les antécédents de diabète, les revascularisations coronariennes antérieures et les antécédents d'insuffisance cardiaque ont donné des valeurs p similaires.
IM : infarctus du myocarde, UHARCR : hospitalisation urgente pour angornécessitant une revascularisation coronarienne
Lanalyse de sensibilité du critère d'évaluation principal (HR 0,72 ; IC à 95 % : 0,56 à 0,93 ; p = 0,01) pour la population prédéfinie par protocole (PP) (patients sans écarts majeurs du protocole) et lanalyse en cours de traitement (patients recevant leur thérapie) (HR 0,65 ; IC à 95 %, 0,50 à 0,84 ; p = 0,001) ont également montré des différences statistiquement significatives entre les groupes.
Lanalyse de sous-groupe (par sexe, statut tabagique, antécédents de diabète, antécédents d'hypertension, antécédents d'IM, antécédents d'ICP ou de pontage aorto-coronarien (PAC), antécédents d'accident vasculaire cérébral ou d'accident ischémique transitoire (AIT) et nombre de globules blancs initial (WBC)) du critère dévaluation principal na montré aucun effet dinteraction significatif.
Une analyse en fonction du délai dinitiation du traitement a montré un effet plus important sur le critère principal lorsque les patients étaient recrutés dans l'étude de 0 à 3 jours après l'IM d'index, par rapport à un délai dinitiation de 4 à 7 jours ou de 8 à 30 jours.
L'analyse des critères d'évaluation secondaires a montré que le décès cardiovasculaire, l'arrêt cardiaque, l'IM aigu ou l'accident vasculaire cérébral sont survenus chez 4,7 % des patients traités par Migoutine, contre 5,5 % des patients du groupe placebo (HR 0,85 ; 95 % IC, 0,66 à 1,10, p=0,22) (voir Tableau 4). Les taux de tous les événements observés pour tous les composants du critère d'évaluation principal étaient plus faibles avec Migoutine par rapport au placebo , avec une différence significative pour l'AVC (HR : 0,26 ; IC à 95 %, 0,10 à 0,70 ; p = 0,01) et l'UHARCR (HR : 0,50 ; 95). % IC, 0,31 à 0,81 ; p = 0,005). La mortalité toutes causes, est survenue chez 44 patients au sein de chaque groupe de létude.
Le taux d'événements totaux (premiers et récurrents) sur le critère d'évaluation principal a été réduit de manière significative de 34 % avec Migoutine par rapport au placebo (rapport des taux 0,66 ; IC à 95 %, 0,51 à 0,86, p = 0,002) dans la population de l'étude en ITT.
La population de lanalyse de sécurité dans COLCOT était composée de 4 676 patients, dont 2 330 patients recevant Migoutine et 2 346 patients recevant un placebo. Aucune exposition à la colchicine avant randomisation n'a eu lieu dans cet essai.
Des événements indésirables liés au traitement (TEAE) ont été rapportés chez 16,0 % des patients traités par Migoutine et chez 15,8 % des patients du groupe placebo (voir Tableau 3). Des EIIT graves liés au traitement médicamenteux sont survenus chez 1,1 % des patients du groupe Migoutine et chez 0,8 % des patients du groupe placebo. La diarrhée et les nausées ont été rapportées avec une incidence plus élevée dans le groupe Migoutine (8,0 % et 1,2 % respectivement), par rapport au groupe placebo (7,3 % et 0,6 % respectivement) (voir Tableau 3).
Table 3 TEAE associés par classe de systèmes d'organes MedDRA et terme préféré chez ≥ 1 % des patients de l'essai COLCOT (population de sécurité)
|
|
1 Aucun déséquilibre notable des valeurs de laboratoire spécifiques n'a été observé entre les groupes de traitement.
2 Comprend l'alopécie (0,3 %), la dermatite allergique (0,1 %), l'érythème (0,1 %) et l'éruption prurigineuse (0,1 %), chez les patients traités par la colchicine.
Dans le groupe Migoutine, 16,4 % des patients ont présenté des EIIT graves, contre 17,2 % dans le groupe placebo. Les événements indésirables graves (EIG) les plus fréquemment signalés étaient les douleurs thoraciques (1,6 %) et l'angine de poitrine (1,2 %), avec une incidence similaire dans les groupes. La pneumonie, en tant qu'EIG, est survenue chez 0,9 % des patients traités par la colchicine et 0,4 % des patients traités par placebo.
Au total, 1 227 TEAE gastro-intestinaux (GI) ont été rapportés chez 17,5 % des patients du groupe Migoutine et chez 17,6 % des patients du groupe placebo. Des événements indésirables gastro-intestinaux graves ont été rapportés chez 2,0 % des patients traités par colchicine et 1,5 % de ceux traités par placebo, l'hémorragie gastro-intestinale étant l'événement indésirable le plus fréquemment signalé : 0,3 % dans le groupe colchicine et 0,2 % dans le groupe placebo. Le médicament à l'étude a été arrêté définitivement en raison d'un GI TEAE chez 4,4 % des patients traités par la colchicine et 3,8 % des patients traités par placebo.
Population pédiatrique
L'Agence européenne des médicaments a levé l'obligation de soumettre les résultats des études réalisées avec Migoutine dans tous les sous-groupes de la population pédiatrique pour la prévention des événements cardiovasculaires (voir rubrique 4.2 pour les informations sur l'usage pédiatrique).
5.2. Propriétés pharmacocinétiques
Après son administration par voie orale, la colchicine est soumise à un cycle entéro-hépatique. Elle est rapidement absorbée par le tractus gastro-intestinal. Chez les adultes en bonne santé qui ont reçu une dose unique de Migoutine à jeun par voie orale, la colchicine absorbée atteint une Cmax moyenne d'un peu moins de 2 ng/mL (plage de 0,8 à 3,9 ng/mL) en environ 1,3 heure (plage de 0,75 à 4 heures).
Lorsque Migoutine a été administré avec ou après un repas riche en graisses et en calories dans le cadre d'une étude de biodisponibilité, les résultats ont montré l'absence d'effet alimentaire significatif. Migoutine peut être administré avec ou sans nourriture.
Chez certains sujets, des pics secondaires de colchicine sont observés, survenant entre 3 et 36 heures après l'administration et allant de 39 % à 155 % de la hauteur du pic initial. Ces observations sont attribuées à la sécrétion et à la réabsorption intestinales et/ou à la recirculation biliaire.
La biodisponibilité absolue serait d'environ 45 %.
Distribution
Le volume de distribution apparent moyen chez les jeunes volontaires sains est d'environ 5 à 8 L/kg.
La liaison de la colchicine aux protéines sériques est faible, 39 ± 5 %, principalement à l'albumine, quelle que soit la concentration.
Après réabsorption, la colchicine est rapidement éliminée du plasma et distribuée dans divers tissus. La colchicine se trouve en concentrations élevées dans les leucocytes, les reins, le foie et la rate et, par conséquent, son accumulation dans ces tissus peut entraîner une toxicité. La colchicine est rapidement distribuée dans les leucocytes périphériques et les concentrations dans ces cellules peuvent dépasser celles du plasma.
Biotransformation
La colchicine est en partie acétylée dans le foie et est lentement métabolisée dans d'autres tissus. Il est déméthylé en deux métabolites principaux, la 2-O-déméthylcolchicine et la 3-O-déméthylcolchicine (respectivement 2-et 3-DMC) et en un métabolite mineur, la 10-O-déméthylcolchicine (également connue sous le nom de colchicéine). Des études in vitro utilisant des microsomes hépatiques humains ont montré que le CYP3A4 est impliqué dans le métabolisme de la colchicine en 2-et 3-DMC. Les taux plasmatiques de ces métabolites sont minimes (moins de 5 % du médicament d'origine). La colchicine est un substrat de la P-gp. La co-administration avec des inhibiteurs puissants de la P-gp et/ou des inhibiteurs puissants du CYP3A4 augmentera l'exposition à la colchicine, ce qui peut entraîner une toxicité induite par la colchicine, voire mortelle (voir rubrique 4.5).
Élimination
Chez des volontaires sains, 40 à 65 % de 1 mg de colchicine administrée par voie orale ont été retrouvés sous forme inchangée dans l'urine. La recirculation entérohépatique et l'excrétion biliaire joueraient également un rôle dans l'élimination de la colchicine. Après plusieurs doses orales (0,6 mg deux fois par jour), la demi-vie d'élimination moyenne chez les jeunes volontaires sains (âge moyen de 25 à 28 ans) est de 26,6 à 31,2 heures.
Élimination extracorporelle
La colchicine n'est pas éliminée par hémodialyse.
Linéarité/non-linéarité
Chez les adultes en bonne santé, la colchicine présente une pharmacocinétique linéaire dans une plage de doses de 0,5 à 1,5 mg.
Insuffisance rénale
Aucune étude pharmacocinétique dédiée n'a été menée avec Migoutine chez des patients présentant divers degrés d'insuffisance rénale. Un rapport publié décrit l'élimination de la colchicine (1 mg) chez de jeunes hommes et femmes adultes atteints de fièvre méditerranéenne familiale (FMF) qui avaient une fonction rénale normale ou une insuffisance rénale terminale nécessitant une dialyse. Les patients atteints d'insuffisance rénale terminale (IRT) présentaient une clairance de la colchicine 75 % inférieure (0,17 contre 0,73 L/h/kg) et une demi-vie d'élimination plasmatique prolongée (18,8 heures contre 4,4 heures) par rapport aux sujets atteints de FMF et avec une fonction rénale normale. Dans une autre étude, 8 sujets sains ayant une fonction rénale normale, 8 sujets présentant chacun une insuffisance rénale légère, modérée ou sévère et 8 sujets atteints dIRT ont reçu une dose unique de 0,6 mg de colchicine avant de recevoir, puis de nouveau après, une hémodialyse. Les résultats ont montré que lexposition à la colchicine était similaire chez les sujets présentant une fonction rénale normale, une insuffisance légère ou une IRT avant et pendant lhémodialyse (24,7 à 31,7 ng·h/mL), mais était jusquà deux fois plus élevée chez les sujets présentant une atteinte dune insuffisance rénale modérée ou sévère (48,9 et 48,0 ng·h/mL, respectivement). Une très petite quantité de la dose de colchicine (moyenne de 5,2 %) a été récupérée dans le dialysat.
Insuffisance hépatique
Aucune étude pharmacocinétique dédiée à Migoutine n'a été menée chez des patients présentant divers degrés d'insuffisance hépatique. Les rapports publiés sur la pharmacocinétique de la colchicine IV chez les patients atteints d'une maladie hépatique chronique sévère, ainsi que chez ceux atteints d'une cirrhose biliaire alcoolique ou primitive et d'une fonction rénale normale suggèrent une grande variabilité inter-patients. Chez certains sujets atteints de cirrhose légère à modérée, la clairance de la colchicine est significativement réduite et la demi-vie plasmatique prolongée par rapport aux sujets sains. Chez les sujets atteints de cirrhose biliaire primitive, aucune tendance cohérente n'a été notée. Aucune donnée pharmacocinétique n'est disponible pour les patients présentant une insuffisance hépatique sévère (Child-Pugh C).
5.3. Données de sécurité préclinique
Toxicologie générale : Aucune étude de toxicologie générale n'a été menée avec Migoutine. Tout surdosage en colchicine est potentiellement mortel.
Cancérogénicité : Des études de deux ans ont été menées chez la souris et le rat pour évaluer le potentiel cancérigène de la colchicine. Aucun signe de tumorigénicité liée à la colchicine n'a été observé chez la souris ou le rat à des doses orales de colchicine allant jusqu'à 3 mg/kg/jour et 2 mg/kg/jour, respectivement.
Génotoxicité : La colchicine s'est révélée mutagène dans le test de mutation inverse bactérienne. Dans un test d'aberration chromosomique sur des globules blancs humains en culture, le traitement à la colchicine a entraîné la formation de micronoyaux. Étant donné que des études publiées ont démontré que la colchicine induit une aneuploïdie à partir du processus de non-disjonction mitotique sans modifications structurelles de l'ADN, la colchicine n'est pas considérée comme clastogène, bien que des micronoyaux se forment.
Toxicologie de la reproduction et du développement : des études non cliniques publiées ont démontré que la perturbation de la formation des microtubules induite par la colchicine affecte la méiose et la mitose. Des études sur la reproduction ont également signalé une morphologie anormale des spermatozoïdes et une diminution du nombre de spermatozoïdes chez les mâles, ainsi que des interférences avec la pénétration des spermatozoïdes, la deuxième division méiotique et le clivage normal chez les femelles exposées à la colchicine. La colchicine administrée à des animaux gravides a entraîné la mort ftale et une tératogénicité. Ces effets étaient dépendants de la dose, le moment de l'exposition étant crucial pour les effets sur le développement embryoftal. Les doses non cliniques évaluées étaient généralement supérieures à une dose thérapeutique humaine équivalente, mais les marges de sécurité pour la toxicité sur la reproduction et le développement n'ont pas pu être déterminées.
Povidone K30
Carboxyméthylamidon sodique (type A)
Sucre compressible : maltodextrine, saccharose, sirop de sucre inverti
Stéarate de magnésium (E572)
Pelliculage du comprimé :
Opadry II Vert 85F91162 :
Alcool polyvinylique partiellement hydrolysé (E1203)
Polyéthylèneglycol MW 3350/Macrogol 4000
Dioxyde de titane (E171)
Talc (E553b)
Oxyde de fer jaune (E172)
Laque aluminique bleu brillant FCF (E133)
Dispersion de copolymère dacide méthacrylique méthacrylate de méthyle (1:2)
Plasacryl T20 :
Monostéarate de glycéryle
Citrate de triéthyle
Polysorbate 80
3 ans
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
Les comprimés seront conditionnés dans des flacons en PEHD, avec un sachet tamis moléculaire, de 30, 90 et 100 comprimés.
Toutes les présentations ne peuvent pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
Pharmascience International Limited
Lampousas 1, 1095,
Nicosia
Chypre
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 302 938 0 5 : 30 comprimés en flacon (PEHD).
· 34009 551 014 3 7 : 90 comprimés en flacon (PEHD).
· 34009 551 014 4 4 : 100 comprimés en flacon (PEHD).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
