RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ANSM - Mis à jour le : 02/01/2025
Ig VENA 50 g/L, solution pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Immunoglobuline humaine normale (IgIV).
1 mL de solution contient :
Immunoglobuline humaine normale ......................................................................................... 50 mg
(pureté : au moins 95 % d'IgG)
Un flacon de 20 mL contient : 1 g d'immunoglobuline humaine normale
Un flacon de 50 mL contient : 2,5 g d'immunoglobuline humaine normale
Un flacon de 100 mL contient : 5 g d'immunoglobuline humaine normale
Un flacon de 200 mL contient : 10 g d'immunoglobuline humaine normale
Répartition des sous-classes d'IgG (valeurs approximatives) :
IgG1 62,1 %
IgG2 34,8 %
IgG3 2,5 %
IgG4 0,6 %
La teneur maximale en IgA est de 50 microgrammes/mL.
Produit à partir de plasma de donneurs humains.
Excipient à effet notoire :
Le produit contient 100 mg de maltose par mL.
Pour la liste complète des excipients, voir rubrique 6.1.
La solution doit être limpide ou légèrement opalescente, incolore ou jaune pâle.
4.1. Indications thérapeutiques
Traitement substitutif chez les adultes, les enfants et les adolescents (0-18 ans) atteints de :
· Déficits immunitaires primitifs (DIP) avec altération de la production danticorps.
· Déficits immunitaires secondaires (DIS) chez les patients souffrant dinfections sévères ou récurrentes, en échec dun traitement antimicrobien et ayant, soit un défaut de production danticorps spécifiques (DPAS)* avéré, soit un taux dIgG sériques < 4 g/l.
* DPAS = incapacité à augmenter dau moins 2 fois le titre danticorps IgG dirigés contre les antigènes polysaccharidiques et polypeptidiques des vaccins anti-pneumococciques.
Traitement immunomodulateur chez les adultes, les enfants et les adolescents (0- 18 ans) atteints de :
· Thrombocytopénie immune primaire (TIP), chez les patients présentant un risque hémorragique important ou avant une intervention chirurgicale pour corriger le taux de plaquettes.
· Syndrome de Guillain Barré.
· Maladie de Kawasaki (en association avec lacide acétylsalicylique ; voir rubrique 4.2).
· Polyradiculonévrite inflammatoire démyélinisante chronique (PIDC).
· Neuropathie motrice multifocale (NMM).
4.2. Posologie et mode d'administration
Posologie
La dose et le schéma posologique varient en fonction de l'indication.
La dose doit être personnalisée pour chaque patient en fonction de la réponse clinique. La dose basée sur le poids corporel peut nécessiter une adaptation chez les patients en sous-poids ou en surpoids.
Les posologies suivantes sont données à titre indicatif.
Traitement substitutif des déficits immunitaires primitifs
La posologie doit permettre datteindre un taux résiduel dIgG (mesuré avant la perfusion suivante) dau moins 6 g/l ou dans la fourchette de référence pour la tranche dâge. Trois à six mois sont nécessaires après le début du traitement pour atteindre léquilibre (taux dIgG à létat déquilibre). La dose initiale recommandée est de 0,4 à 0,8 g/kg en dose unique suivie dau moins 0,2 g/kg toutes les 3 à 4 semaines.
La dose nécessaire pour atteindre un taux de résiduel dIgG de 6 g/l est de lordre de 0,2 à 0,8 g/kg/mois.
Lintervalle entre les doses varie de 3 à 4 semaines lorsque létat déquilibre est atteint.
Les taux résiduels dIgG doivent être mesurés et évalués en fonction de lincidence des infections.
Pour réduire le taux dinfections bactériennes, il peut être nécessaire daugmenter la dose afin datteindre des taux résiduels plus élevés.
Déficits immunitaires secondaires (tels que définis à la rubrique 4.1)
La dose recommandée est de 0,2 à 0,4 g/kg toutes les 3 à 4 semaines.
Les taux résiduels dIgG doivent être mesurés et évalués en fonction de lincidence des infections. La dose doit être ajustée comme nécessaire pour lobtention dune protection optimale contre les infections et une augmentation de la dose peut être nécessaire chez les patients présentant une infection persistante ; une diminution de la dose peut être envisagée lorsque le patient ne développe pas dinfection.
Thrombocytopénie immune primaire
Il existe deux schémas de traitements alternatifs :
· 0,8 à 1 g/kg le premier jour ; cette dose peut être répétée une fois dans les trois jours ;
· 0,4 g/kg par jour pendant 2 à 5 jours.
Le traitement peut être répété en cas de rechute.
Syndrome de Guillain Barré
0,4 g/kg/jour pendant 5 jours (le traitement peut être répété en cas de rechute).
Maladie de Kawasaki
2,0 g/kg administrés en dose unique. Les patients doivent recevoir un traitement associé par lacide acétylsalicylique.
Polyradiculonévrite inflammatoire démyélinisante chronique (PIDC)
Dose initiale : 2 g/kg répartie sur 2 à 5 jours consécutifs.
Doses dentretien : 1 g/kg sur 1 à 2 jours consécutifs toutes les 3 semaines.
Leffet du traitement doit être évalué après chaque cycle ; si aucun effet nest observé après 6 mois, le traitement doit être interrompu.
Si le traitement est efficace, la possibilité dun traitement à long terme devra être laissée à la discrétion des médecins en fonction de la réponse du patient et du maintien de la réponse. La posologie et les intervalles dadministration peuvent être adaptés au cas par cas en fonction de lévolution de la maladie.
Neuropathie motrice multifocale (NMM)
Dose initiale : 2 g/kg répartie sur 2 à 5 jours consécutifs.
Doses dentretien : 1 g/kg toutes les 2 à 4 semaines ou 2 g/kg toutes les 4 à 8 semaines.
Leffet du traitement doit être évalué après chaque cycle ; si aucun effet nest observé après 6 mois, le traitement doit être interrompu.
Si le traitement est efficace, la possibilité dun traitement à long terme devra être laissée à la discrétion des médecins en fonction de la réponse du patient et du maintien de la réponse. La posologie et les intervalles dadministration peuvent être adaptés au cas par cas en fonction de lévolution de la maladie.
Les doses recommandées sont résumées dans le tableau suivant :
|
Indication |
Dose |
Fréquence des injections |
|
Traitement substitutif |
|
|
|
Déficits immunitaires primitifs |
Dose initiale : 0,4 0,8 g/kg Dose dentretien : 0,2 0,8 g/kg |
Toutes les 3 à 4 semaines |
|
Déficits immunitaires secondaires (tels que définis à la rubrique 4.1) |
0,2 - 0,4 g/kg |
Toutes les 3 à 4 semaines |
|
Traitement immunomodulateur : |
|
|
|
Thrombocytopénie immune primaire |
0,8 - 1 g/kg |
Le jour 1, avec possibilité de répéter le traitement dans les 3 jours |
|
ou |
||
|
0,4 g/kg/jour |
Pendant 2 à 5 jours |
|
|
Syndrome de Guillain Barré |
0,4 g/kg/jour |
Pendant 5 jours |
|
Maladie de Kawasaki |
2 g/kg |
En dose unique en association avec lacide acétylsalicylique |
|
Polyradiculonévrite inflammatoire démyélinisante chronique (PIDC) |
Dose initiale : 2 g/kg |
En plusieurs doses réparties sur 2 à 5 jours |
|
|
Dose dentretien : 1 g/kg |
Toutes les 3 semaines sur 1 à 2 jours |
|
Neuropathie motrice multifocale (NMM) |
Dose initiale : 2 g/kg |
De 2 à 5 jours consécutifs |
|
Dose dentretien : 1 g/kg ou |
Toutes les 2 à 4 semaines ou |
|
|
2 g/kg |
Toutes les 4 à 8 semaines sur 2 à 5 jours |
|
Population pédiatrique
La posologie chez les enfants et les adolescents (0-18 ans) n'est pas différente de celle des adultes, étant donné que la posologie pour chaque indication étant calculée en fonction du poids corporel et ajustée en fonction du résultat clinique des conditions susmentionnées.
Insuffisance hépatique
Il nexiste pas de données indiquant quun ajustement de la dose est nécessaire.
Insuffisance rénale
Aucun ajustement de la dose nest nécessaire, sauf sil est cliniquement justifié, voir rubrique 4.4.
Sujets âgés
Aucun ajustement de la dose nest nécessaire, sauf sil est cliniquement justifié, voir rubrique 4.4
Polyradiculonevrite Inflammatoire Démyélinisante Chronique (PIDC)
En raison de la rareté de la maladie et, par conséquent, du nombre limité total de patients, l'expérience dans l'utilisation d'immunoglobulines par voie intraveineuse chez les enfants atteints de PIDC est limitée ; par conséquent, on ne dispose que des données de la littérature scientifique. Cependant, les données publiées convergent lorsqu'il s'agit de montrer que le traitement avec IgIV est efficace de la même manière chez les enfants et les adultes, conformément à ce qui se passe pour les indications reconnues pour les IgIV.
Mode dadministration
Utilisation par voie intraveineuse.
L'immunoglobuline humaine normale doit être perfusée par voie intraveineuse à une vitesse initiale de 0,46 - 0,92 mL/kg/h (10 à 20 gouttes par minute) pendant 20 à 30 minutes. Voir rubrique 4.4. En cas de survenue dun effet indésirable, le débit de perfusion doit être diminué ou la perfusion arrêtée. Si la perfusion est bien tolérée, le débit peut être accéléré progressivement jusquà un maximum de 1,85 mL/kg/h (40 gouttes par minute).
Chez les patients souffrant de DIP qui tolèrent bien la vitesse de perfusion de 0,92 mL/kg/h, la vitesse d'administration peut être augmentée progressivement à 2 mL/kg/h, 4 mL/kg/h, jusqu'à un maximum de 6 mL/kg/h, toutes les 20 à 30 minutes et seulement si le patient tolère bien la perfusion.
En général, le dosage et la vitesse de perfusion doivent être adaptés de manière singulière aux besoins du patient. En fonction du poids corporel, du dosage et de l'apparition d'effets indésirables, le patient peut ne pas atteindre la vitesse de perfusion maximum. En cas d'effet indésirable, la perfusion doit être immédiatement arrêtée et doit être reprise en appliquant la vitesse qui convient le mieux au patient.
Voir la rubrique 6.6
Populations particulières
Chez les patients pédiatriques (0-18 ans) et les personnes âgées (> 64 ans), la vitesse initiale d'administration doit être de 0,46 - 0,92 mL/kg/h (10 à 20 gouttes/minute) pendant 20 à 30 minutes. Si elle est bien tolérée et en fonction des conditions cliniques du patient, la vitesse d'administration peut être progressivement augmentée jusqu'à un maximum de 1,85 mL/kg/h (40 gouttes/ minutes).
Patients présentant un déficit sélectif en IgA ayant développé des anticorps anti-IgA, car ladministration dun produit contenant des IgA peut provoquer une anaphylaxie
4.4. Mises en garde spéciales et précautions d'emploi
Ce produit contient 100 mg de maltose par mL comme excipient. L'interférence du maltose avec les tests de la glycémie peut conduire à une surestimation des taux de glucose et, par conséquent, à une administration d'insuline inadéquate, qui peut entraîner un état d'hypoglycémie avec risque de vie et un décès du patient. En outre, les cas réels d'hypoglycémie peuvent ne pas être traités si l'état hypoglycémique est masqué par des taux de glucose faussement élevés. Pour plus de détails, voir la rubrique 4.5. Pour l'insuffisance rénale aiguë, voir ci-dessous.
Ce médicament contient environ 3 mmol/litre (ou 69 mg/litre) de sodium. Ceci doit être pris en compte pour les patients suivant un régime alimentaire à base de sodium contrôlé.
Traçabilité
Afin daméliorer la traçabilité des médicaments biologiques, le nom de marque et le numéro de lot du produit administré doivent être clairement enregistrés.
Précautions demploi
Des complications potentielles graves peuvent souvent être évitées en s'assurant :
· que les patients ne sont pas sensibles à l'immunoglobuline humaine normale en administrant le produit lentement au début (avec une vitesse de perfusion comprise entre 0,46 - 0,92 mL/kg/h) ;
· que les patients sont attentivement contrôlés pour n'importe quel symptôme au cours de la période de perfusion. En particulier, les patients recevant de l'immunoglobuline humaine normale pour la première fois, les patients qui ont changé de type de produit à base d'IgIV et les patients pour lesquels un long laps de temps s'est écoulé depuis la perfusion précédente doivent être surveillés au cours de la première perfusion et au cours de la première heure après la première perfusion, afin d'identifier d'éventuels signes d'effets indésirables. Tous les patients devront être surveillés pendant au moins 20 minutes après l'administration.
Chez tous les patients, ladministration dIgIV exige :
· une hydratation appropriée avant le début de la perfusion dIgIV,
· le contrôle du volume urinaire,
· le contrôle du taux de créatinine sérique,
· la non-utilisation concomitante de diurétiques de lanse (voir la rubrique 4.5)
En cas deffets indésirables, le débit dadministration doit être diminué ou la perfusion arrêtée. Le traitement requis dépend de la nature et de la sévérité de leffet indésirable.
Réaction à la perfusion
Certains effets indésirables (ex. céphalées, bouffées vasomotrices, frissons, myalgies, sibilances, tachycardie, douleur au bas du dos, nausées et hypotension) peuvent être liés au débit de perfusion. Le débit de perfusion recommandé à la rubrique 4.2 doit être strictement respecté. Les patients doivent être étroitement surveillés et attentivement observés pendant toute la durée de la perfusion afin de détecter tout symptôme.
Des effets indésirables peuvent survenir plus fréquemment :
· chez les patients qui reçoivent des immunoglobulines humaines normales pour la première fois ou, dans de rares cas, en cas de changement dimmunoglobulines humaines normales ou de long délai depuis la perfusion précédente ;
· chez les patients présentant une infection non traitée ou une inflammation chronique.
Hypersensibilité
Les réactions dhypersensibilité sont rares.
Une anaphylaxie peut survenir chez les patients :
· présentant des anticorps anti-IgA avec IgA indétectables ;
· qui avaient toléré un traitement antérieur par des immunoglobulines humaines normales.
En cas de choc, le traitement habituel dun choc doit être instauré.
Thromboembolie
Cliniquement, lexistence dun lien est reconnue entre ladministration dIgIV et des événements thromboemboliques tels quinfarctus du myocarde, accident vasculaire cérébral (y compris ictus), embolie pulmonaire et thrombose veineuse profonde. Ces événements sont probablement liés à une élévation relative de la viscosité sanguine provoquée par un apport important en immunoglobulines chez les patients à risque. Toutes les précautions doivent être prises en cas de prescription et dadministration dIgIV chez les patients obèses, chez les patients présentant des facteurs de risque thrombotique préexistants (âge avancé, hypertension, diabète sucré, et antécédents de maladie vasculaire ou dépisodes thrombotiques, patients présentant une thrombophilie acquise ou héréditaire, patients immobilisés durant des périodes prolongées, patients sévèrement hypovolémiques et patients atteints de maladies entraînant une augmentation de la viscosité sanguine).
Chez les patients présentant un risque dévénement indésirable thromboembolique, les IgIV doivent être administrées à une dose et un débit de perfusion les plus faibles possibles.
Insuffisance rénale aiguë
Des cas dinsuffisance rénale aiguë ont été rapportés chez des patients recevant un traitement par IgIV. Dans la plupart des cas, des facteurs de risque ont été identifiés, par exemple insuffisance rénale préexistante, diabète sucré, hypovolémie, surpoids, administration concomitante de médicaments néphrotoxiques ou âge supérieur à 65 ans.
Les paramètres rénaux doivent être évalués avant la perfusion d'IgIV, en particulier chez les patients considérés comme potentiellement à risque de développer une insuffisance rénale aiguë, et à nouveau à des intervalles appropriés. Chez les patients présentant un risque dinsuffisance rénale aiguë, les IgIV doivent être administrées à une dose et à un débit de perfusion les plus faibles possibles. Dans le cas d'altérations de la fonction rénale, il faut envisager d'interrompre le traitement par IgIV.
Même si des cas de dysfonction rénale et d'insuffisance rénale aiguë ont été associés à l'utilisation de nombreux médicaments à base d'IgIV contenant divers excipients comme le saccharose, le glucose et le maltose, ceux qui contiennent du saccharose comme stabilisateur représentent un pourcentage très élevé du nombre total. Chez les patients à risque, on peut envisager l'utilisation de médicaments à base d'IgIV qui ne contiennent pas ces excipients. Ig VENA contient du maltose (voir les excipients à la rubrique 6.1).
Syndrome de méningite aseptique (SMA)
Le syndrome de méningite aseptique peut se manifester en association avec le traitement avec IgIV. Le syndrome débute généralement dans les quelques heures à 2 jours suivant ladministration dIgIV. Les études sur le liquide céphalo-rachidien sont souvent positives pour des pléiocytoses allant jusqu'à plusieurs milliers de cellules par mm3, surtout des granulocytes et des taux élevés en protéines, jusqu'à plusieurs centaines de mg/dl.
Un SMA peut survenir plus fréquemment en cas de traitement par IgIV à dose élevée (2 g/kg).
Chez les patients présentant de tels signes et symptômes, un examen neurologique approfondi et une analyse du LCR doivent être réalisés afin dexclure dautres causes de méningite.
Larrêt du traitement par IgIV a permis une rémission du SMA en quelques jours, sans séquelles.
Anémie hémolytique
Les produits à base d'IgIV peuvent contenir des anticorps spécifiques à des groupes sanguins qui peuvent agir comme des hémolysines et induire un recouvrement in vivo des globules rouges par les immunoglobulines, ce qui provoque une réaction antiglobulinique directe positive (test de Coombs) et, dans de rares cas, une hémolyse. L'anémie hémolytique peut se développer à la suite du traitement avec IgIV en raison d'une séquestration accrue des globules rouges. Les patients traités par IgIV doivent être surveillés afin de déceler tous signes cliniques et symptômes dhémolyse. (Voir rubrique 4.8).
Neutropénie/leucopénie
Une diminution transitoire du taux de neutrophiles et/ou des épisodes de neutropénie, parfois sévères, ont été rapportés après un traitement par IgIV. La diminution survient généralement dans les quelques heures ou jours suivant ladministration dIgIV et est spontanément résolutive en 7 à 14 jours.
Syndrome de détresse respiratoire aiguë post-transfusionnel (TRALI)
Chez les patients recevant des IgIV quelques cas ddème pulmonaire aigu non cardiogénique (syndrome de détresse respiratoire aiguë post-transfusionnel, TRALI) ont été rapportés. Le TRALI est caractérisé par hypoxie grave, dyspnée, tachypnée, cyanose, fièvre et hypotension. Les symptômes du TRALI apparaissent généralement pendant une perfusion ou dans les 6 heures suivant la fin de la perfusion, souvent en 1 à 2 heures. Par conséquent, les patients traités par IgIV doivent être surveillés et la perfusion dIgIV doit être immédiatement arrêtée en cas de survenue deffets indésirables pulmonaires. Le TRALI est une affection susceptible dengager le pronostic vital nécessitant une prise en charge immédiate dans une unité de soins intensifs.
Interférence avec des tests sérologiques
Après la perfusion d'immunoglobuline, l'augmentation transitoire de différents anticorps transférés passivement dans le sang du patient peut donner des résultats faussement positifs des tests sérologiques.
La transmission passive d'anticorps dirigés contre les antigènes érythrocytaires, par exemple A, B, D peut interférer avec certains tests sérologiques pour les anticorps des globules rouges, par exemple le test antiglobuline (TAD, test de Coombs direct).
Agents transmissibles
Les mesures standards pour prévenir les infections résultant de l'utilisation de médicaments préparés à partir de sang ou de plasma humain incluent la sélection des donneurs, le dépistage sur les dons individuels et sur les pools de plasma pour des marqueurs spécifiques d'infection et l'inclusion d'étapes de fabrication efficaces pour l'inactivation / élimination des virus.
Malgré cela, la possibilité de transmission dagents infectieux lors de ladministration de médicaments préparés à partir de sang ou de plasma humain ne peut être totalement exclue. Ceci s'applique aussi aux virus et à d'autres agents pathogènes émergents ou inconnus.
Les mesures adoptées sont considérées comme efficaces vis-à-vis des virus enveloppés, tels que le VIH, le VHB et le VHC, ainsi que des virus non enveloppés comme le VHA.
Les mesures adoptées présentent une valeur limitée contre les virus sans enveloppe lipidique tels que le parvovirus B19.
Il existe une expérience clinique rassurante concernant l'absence de transmission de l'hépatite A et du parvovirus B19 avec les immunoglobulines et on peut supposer que le contenu d'anticorps apporte une contribution importante à la sécurité virale.
Il est fortement recommandé quà chaque administration dIg VENA, le nom et le numéro de lot soient enregistrés, de façon à garantir la traçabilité entre le patient et le lot utilisé.
Population pédiatrique
Des cas de glycosurie ont été signalés chez les enfants après l'administration d'Ig VENA. Ces événements sont généralement légers et transitoires, sans signes cliniques.
Ig VENA contient 100 mg de maltose par mL comme excipient. Dans les tubules rénaux, le maltose est hydrolysé en glucose, qui est réabsorbé et n'est généralement excrété dans les urines que dans une faible mesure. La réabsorption du glucose est un mécanisme dépendant de l'âge. L'augmentation transitoire de maltose dans le plasma peut dépasser la capacité du rein de réabsorber le sucre et entraîner un test positif pour le glucose dans les urines.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Vaccins à virus vivants atténués
L'administration d'immunoglobulines peut compromettre au cours d'une période d'au moins 6 semaines et jusqu'à 3 mois l'efficacité des vaccins à virus vivants atténués tels que la rougeole, la rubéole, la parotidite et la varicelle. Après l'administration de ce produit, il faut que sécoule un intervalle d'au moins trois mois avant la vaccination avec des vaccins à virus vivants atténués. Dans le cas de la rougeole, cet affaiblissement de la réponse peut se prolonger jusqu'à un an. Par conséquent, chez les patients qui reçoivent le vaccin de la rougeole, il faut que le niveau des anticorps soit vérifié.
Diurétiques de lanse
Lutilisation concomitante de diurétiques de lanse doit être évitée.
Test de la glycémie
Certains systèmes de mesure de la glycémie (par exemple, ceux basés sur le glucose déshydrogénase pyrroloquinoléine quinone (GDH-PQQ) ou sur la méthode colorimétrique de la glucose-oxydoréductase) reconnaissent faussement le maltose (100 mg/mL) contenu dans Ig VENA comme étant du glucose. Cela peut se traduire par une lecture de taux de glycémie faussement élevés pendant la perfusion et pendant une période d'environ 15 heures après la fin de la perfusion et, par conséquent, par une administration inadéquate d'insuline, provoquant un danger pour la vie ou même une hypoglycémie fatale. En outre, les cas réels d'hypoglycémie peuvent ne pas être traités si l'état hypoglycémique est masqué par des taux de glucose faussement élevés. En conséquence, au cours de l'administration d'Ig VENA ou d'autres produits parentéraux contenant du maltose, la mesure de la glycémie doit être réalisée selon des méthodes spécifiques pour le glucose.
Le mode d'emploi du système de mesure de la glycémie, y compris celui des bandelettes réactives, doit être soigneusement vérifié pour déterminer si le système répond bien à une utilisation chez les patients traités avec des produits parentéraux contenant du maltose.
En cas de doute, contactez le producteur du système de mesure afin de déterminer le caractère approprié d'une utilisation faite en concomitance avec l'usage de produits parentéraux contenant du maltose.
Population pédiatrique
Bien que des études spécifiques n'aient pas été menées à bien en ce qui concerne l'interaction dans la population pédiatrique, on ne prévoit pas de différences par rapport aux patients adultes.
4.6. Fertilité, grossesse et allaitement
Grossesse
L'innocuité de ce médicament au cours de la grossesse n'a pas été établie par des études cliniques contrôlées et il doit donc être utilisé avec prudence chez les femmes enceintes ou allaitant. Il a été démontré que les produits à base d'IgIV traversent le placenta de manière croissante au cours du troisième trimestre de la grossesse.
L'expérience clinique avec les immunoglobulines suggère qu'aucun effet nocif n'est attendu sur le déroulement de la grossesse, ni sur le ftus et le nouveau-né.
Les immunoglobulines sont excrétées dans le lait maternel. Aucun effet indésirable chez les nouveau-nés/nourrissons allaités nest attendu.
Fertilité
L'expérience clinique avec les immunoglobulines suggère qu'aucun effet nocif n'est attendu sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets indésirables causés par les immunoglobulines humaines normales (par ordre décroissant de fréquence) comprennent (voir également rubrique 4.4) :
· frissons, céphalées, étourdissements, fièvre, vomissements, réactions allergiques, nausées, arthralgies, chute de la pression artérielle et douleurs lombaires d'intensité modérée ;
· réactions hémolytiques réversibles, en particulier chez les patients des groupes sanguins A, B et AB et (rarement), anémie hémolytique nécessitant une transfusion ;
· (rarement), chute soudaine de la pression artérielle et, dans certains cas isolés, choc anaphylactique même lorsque le patient n'a pas manifesté de réactions dhypersensibilité au cours d'administrations précédentes ;
· (rarement), réactions cutanées transitoires (y compris lupus érythémateux cutané - fréquence indéterminée) ;
· (très rarement), réactions thromboemboliques telles quinfarctus du myocarde, accident vasculaire cérébral, embolie pulmonaire, thrombose veineuse profonde ;
· cas de méningite aseptique réversible ;
· cas daugmentation de la créatinine et/ou dinsuffisance rénale aiguë ;
· cas de syndrome de détresse respiratoire aiguë post-transfusionnel (TRALI).
La sécurité d'Ig VENA a été évaluée dans quatre études cliniques au cours desquels a été administré un total de 1.189 perfusions. L'étude sur la PIDC a porté sur 24 patients souffrant de Polyneuropathie Démyélinisante Inflammatoire Chronique (PIDC), qui ont reçu Ig VENA, avec un total de 840 perfusions administrées. Dans l'étude clinique sur les Déficits immunitaires primitifs 16 patients présentant un Déficit immunitaires primaire (DIP) ont été recrutés et ont reçu un total de 145 perfusions. L'étude sur la TIP a concerné 15 patients présentant une Thrombocytopénie Immune Primaire (TIP), avec un total de 80 perfusions administrées. Dans l'étude ID/TIP, 43 patients présentant une Immunodéficience (ID) ou une Thrombocytopénie Immune Primaire (TIP) ont été recrutés et ont reçu un total de 124 perfusions.
Liste tabulée des effets indésirables
Le tableau suivant a été établi selon le classement des systèmes d'organes MedDRA (SOC et terme préférentiel).
Le tableau 1 présente les effets indésirables survenus au cours des études cliniques et le tableau 2 présente les effets indésirables rapportés dans l'expérience post-commercialisation.
Les fréquences ont été évaluées selon les conventions suivantes : très fréquent (≥1/10) ; fréquent (≥1/100, <1/10) ; peu fréquent (≥1/1,000, <1/100) ; rare (≥1/10,000, <1/1,000) et très rare (<1/10,000), fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles).
Les fréquences des effets indésirables observés dans les études cliniques sont basées sur un pourcentage par rapport au nombre de perfusions (nombre total de perfusions : 1.189).
Pour les effets indésirables observés après la mise sur le marché la fréquence de signalement est définie comme indéterminée. Ces effets sont rapportés de manière spontanée et proviennent dune population dont les dimensions ne sont pas connues, il nest donc pas possible den estimer la fréquence.
Source de la base de données sur la sécurité (par exemple, à partir d'études cliniques, d'études de sécurité post-autorisation et/ou de rapports spontanés)
|
Tableau 1 Fréquence des effets indésirables identifiés au cours des études cliniques |
|||
|
Classes de Systèmes d'Organes selon MedDRA (SOC) |
Effets indésirables |
Fréquence par patient |
Fréquence de la perfusion |
|
Affections du système nerveux |
Céphalée, somnolence |
Fréquent |
Rare |
|
Affections gastro-intestinales |
Nausée |
Fréquent |
Rare |
|
Affections musculo-squelettiques et systémiques |
Douleur au dos |
Fréquent |
Peu fréquent |
|
Myalgie |
Fréquent |
Rare |
|
|
Troubles généraux et anomalies au site d'administration |
Asthénie, fatigue, pyrexie |
Fréquent |
Rare |
|
Tableau 2 Effets indésirables rapportés durant l'expérience post-commercialisation |
|
||
|
Classes de Systèmes d'Organes selon MedDRA (SOC) |
Effets indésirables |
Fréquence par patient |
Fréquence de la perfusion |
|
Infection et infestations |
Méningite aseptique |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections du système hématologique et lymphatique |
Hémolyse, anémie hémolytique |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections du système immunitaire |
Choc anaphylactique, hypersensibilité |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections psychiatriques |
Confusion mentale |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections du système nerveux |
Accident vasculaire cérébral, céphalée, étourdissement, tremblement, paresthésie |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections cardiaques |
Infarctus du myocarde, cyanose, tachycardie, bradycardie, palpitations |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections vasculaires |
Thrombose veineuse profonde, embolie, hypotension, hypertension, pâleur |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections respiratoires, thoraciques et médiastinales |
Embolie pulmonaire, dème pulmonaire, bronchospasme, dyspnée, toux |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections gastro-intestinales |
Vomissement, diarrhée, nausée, douleur abdominale |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections de la peau et du tissu sous-cutané |
Angio-oedème, urticaire, érythème, dermatite, éruption cutanée, prurit, eczéma, hyperhidrose |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections musculo-squelettiques et systémiques |
Arthralgie, douleur au dos, myalgie, douleur au cou, raideur musculo-squelettique |
Fréquence indéterminée |
Fréquence indéterminée |
|
Affections du rein et des voies urinaires |
Atteinte rénale aiguë |
Fréquence indéterminée |
Fréquence indéterminée |
|
Troubles généraux et anomalies au site d'administration |
Phlébite au site d'injection, pyrexie, frissons, douleur thoracique, dème du visage face, malaise |
Fréquence indéterminée |
Fréquence indéterminée |
|
Investigations |
Baisse de la pression artérielle, augmentation créatinine hématique |
Fréquence indéterminée |
Fréquence indéterminée |
En ce qui concerne les risques liés aux agents transmissibles, consulter la rubrique 4.4.
Population pédiatrique
On prévoit que la fréquence, le type et la gravité des effets indésirables chez les enfants soient équivalents à ceux des adultes.
Une glycosurie transitoire a été observée après l'administration d'Ig VENA. Cet événement peut être dû au maltose contenu dans Ig VENA et à la capacité différente des tubules rénaux de réabsorber le glucose, qui est un mécanisme dépendant de l'âge.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
Un surdosage peut entraîner une hypervolémie et une hyperviscosité, particulièrement chez les patients à risque, y compris les patients âgés ou les patients souffrant d'insuffisance cardiaque ou rénale (voir rubrique 4.4).
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme daction
L'immunoglobuline humaine normale contient principalement des immunoglobulines G (IgG) avec un large spectre d'anticorps dirigés contre les agents infectieux.
L'immunoglobuline humaine normale contient les anticorps anti-IgG présents dans la population normale. En général, elle est préparée à partir d'un pool de plasma provenant d'un minimum de 1.000 dons. Elle présente une répartition de sous-classes d'IgG strictement proportionnelle à celle du plasma humain natif. Des doses appropriées de ce médicament peuvent ramener à des niveaux normaux les taux d'immunoglobulines G anormalement bas.
Le mécanisme d'action pour des indications autres que le traitement substitutif n'est pas totalement élucidé.
Efficacité et sécurité clinique
Quatre études cliniques ont été conduites avec Ig VENA : trois études portant sur l'efficacité et la sécurité chez les patients atteints de déficits immunitaires primitifs (DIP), de Thrombocytopénie Immune Primaire (TIP) et de Polyradiculonévrite Démyélinisante Inflammatoire Chronique (PIDC) ; et une étude sur la sécurité et la tolérance d'Ig VENA, administré avec des vitesses de perfusion croissants chez les patients avec Immunodéficience (ID) ou TIP.
Une étude de phase III, prospective, ouverte, chez des patients présentant des déficits immunitaires primitifs (KB028) a évalué le profil pharmacocinétique d'Ig VENA comme objectif primaire. Les objectifs secondaires étaient l'efficacité thérapeutique en termes de prophylaxie pour les épisodes d'infection et de sécurité et la tolérance à court terme, ont été identifiés comme étant des objectifs secondaires. Quinze patients sur les 16 ayant été recrutés, âgés entre 28 et 60 ans, ont été évalués du point de vue de l'efficacité et traités pendant 24 semaines avec Ig VENA (total de 140 perfusions). Le profil pharmacocinétique d'Ig VENA a montré une demi-vie, raisonnablement en correspondance avec les données figurant dans la littérature scientifique, de 26,4 jours. Un patient a développé une pneumonie après 18 semaines de traitement avec Ig VENA, en ayant, cependant, déjà souffert d'infections pulmonaires graves au cours des 10 années précédentes. Aucune infection grave n'a été signalée chez les autres patients recrutés.
Les données obtenues par l'étude KB028 indiquent qu'Ig VENA est sûr et efficace dans le traitement des déficits immunitaires primitifs.
L'étude TIP (KB027) a été une étude de phase III, ouverte, prospective pour l'évaluation de l'efficacité et la tolérance d'Ig VENA chez des patients adultes atteints de purpura thrombocytopénique idiopathique chronique.
L'objectif principal était l'évaluation de l'augmentation de la numération plaquettaire. Les objectifs secondaires étaient : la diminution des manifestations hémorragiques, la durée de la réponse plaquettaire et l'incidence des effets indésirables. Quinze patients ont reçu une dose totale de 2 g/kg chacun, divisés en 5 perfusions quotidiennes de 400 mg/kg sur des jours consécutifs. Un deuxième cycle de 2 g/kg de poids corporel a été donné à un patient dans les 14 premiers jours. Le nombre total de perfusions administrées a été de 80.
Tous les patients recrutés ont atteint une numération plaquettaire de ≥50x109/L, excepté 1, qui a reçu le deuxième cycle de traitement, mais qui n'a pas atteint la numération plaquettaire fixée comme objectif (taux de réponse de 93,3%, 90% CI de 68,1 à 99,8). Aucun événement indésirable na été signalé.
Les résultats obtenus par l'étude KB027 ont fourni la preuve de la tolérance et de l'efficacité thérapeutique d'Ig VENA chez les patients atteints de TIP.
Dans l'étude de phase III KB057, pour l'évaluation de la tolérance et de la sécurité d'Ig VENA administré à des vitesses croissantes, 43 patients adultes ont été recrutés: 38 avec ID et 5 avec TIP, lesquels ont reçu Ig VENA à des posologies approuvées pour les deux indications.
Trente-sept patients avec ID ont été observés pour 3 perfusions et 1 patient avec ID pour 2 perfusions. Quatre patients atteints de TIP ont reçu la dose prévue pour eux en 2 perfusions quotidiennes, tandis qu'à 1 patient, la dose a été administrée en 3 jours (total des perfusions : 124). À la deuxième perfusion, vingt-huit patients sur les 43 recrutés ont été traités à une vitesse maximum de 8 mL/kg/h ; treize patients sur les 43 recrutés n'ont atteint qu'une vitesse de perfusion maximum de 6 mL/kg/h, car la perfusion était terminée avant que le débit puisse être augmenté à l'étape suivante. Au cours de l'étude clinique, deux patients n'ont pas atteint les 8 mL/kg/h, étant donné qu'ils ont développé 3 réactions indésirables au cours de la perfusion avec des vitesses plus basses.
Les résultats obtenus par l'étude montrent qu'Ig VENA, administré à une vitesse de perfusion croissante, a été bien toléré, aussi bien chez des patients avec ID que chez les patients atteints de TIP, et qu'il était possible d'augmenter la vitesse de perfusion jusqu'à un maximum de 6 mL/kg/h et, chez un nombre limité de patients, jusqu'à 8 mL/kg/h.
Des réactions indésirables ont été signalées chez moins de 10 % des patients avec ID et ont été les réactions qui sont généralement associées à l'administration d'IgIV (par exemple : fièvre, douleur au dos, myalgies, asthénie, somnolence et fatigue).
Aucun effet indésirable grave n'a été signalé, de même qu'aucune réaction locale n'a été signalée au point de perfusion.
Études cliniques réalisées avec Ig VENA chez des patients atteints de Polyradiculonévrite démyélinisante inflammatoire chronique (PIDC) :
L'étude en double-aveugle, contrôlé de phase III sur la tolérance et l'efficacité du traitement à long terme avec de fortes doses d'immunoglobulines par voie intraveineuse par rapport à de fortes doses de méthylprednisolone par voie intraveineuse (IVMP) dans la PIDC (KB034) a été réalisé sur un total de 46 patients adultes atteints de PIDC, randomisés pour recevoir Ig VENA (dose : 2 g/Kg/mois sur 4 jours consécutifs pendant 6 mois) ou IVMP (dose : 2 g/mois sur 4 jours consécutifs pendant 6 mois).
Dix des 21 patients traités avec IVMP (47,6 %) ont achevé les 6 mois de l'étude contre 21/24 patients traités avec Ig VENA (87,5%) (p=0,0085). La probabilité cumulative d'interruption du traitement a été significativement plus élevée avec IVMP qu'avec Ig VENA à 15 jours, 2 mois et 6 mois. Onze patients ont interrompu le traitement avec IVMP : huit en raison d'une aggravation progressive après le début du traitement ou par défaut d'amélioration après deux cycles de traitement (respectivement 5 et 3 patients), l'un en raison d'un effet indésirable (gastrite) (9,1 %) et deux par retrait volontaire (18,2 %). Trois patients ont interrompu le traitement avec Ig VENA à la suite d'une aggravation progressive après le début du traitement ou bien par absence d'amélioration après deux cycles de traitement (respectivement 2 patients et 1 patient). Tous les patients qui ont montré une aggravation ou une absence d'amélioration à la suite du traitement avec IVMP ou IgIV sont passés au traitement alternatif, tandis que les trois patients ayant abandonné IVMP à la suite d'une manifestation indésirable ou d'un retrait volontaire ont refusé de recevoir d'autres traitements.
Dans le tableau qui suit sont résumés les résultats concernant les critères secondaires de l'étude (les écarts statistiquement significatifs sont indiqués en caractères gras) :
|
|
Population Intention To Treat (ITT) |
Population Per Protocol (PP) |
||||
|
Critères secondaires |
IgVENA 10 g/200 mL |
MPIV |
p |
IgVENA 10 g/200 mL |
MPIV |
p |
|
Taux de récidive* |
45.8% (n 11/24) |
52.4% (n 11/21) |
0.7683 |
38.1% (n 8/21) |
0% (n 0/10) |
0.0317 |
|
Échelle MRC [delta (p)] |
+4.7 (0.0078) |
+1.8 (0.1250) |
0.6148 |
+4.0 (0.0469) |
+2.0 (0.5000) |
0.5473 |
|
Échelle INCAT (p) |
0.0004 |
0.1877 |
0.3444 |
0.0057 |
0.2622 |
0.9065 |
|
Seuil vibratoire - malléole médiale droite (p) |
<0.0001 |
0.6515 |
0.0380 |
0.0009 |
0.2160 |
0.4051 |
|
Force du poing droit [delta (p)] |
+19.4 (0.0005) |
+5.4 (0.6169) |
0.0641 |
+16.5 (0.0044) |
+14.7 (0.0156) |
0.5012 |
|
Force du poing gauche [delta (p)] |
+16.9 (0.0011) |
+8.8 (0.1170) |
0.1358 |
+12.7 (0.0014) |
+10.5 (0.0156) |
0.3330 |
|
Durée de parcours 10 mètres [delta (p)] |
-3.2 (0.0025) |
-0.5 (0.2051) |
0.0800 |
-3.5 (0.0043) |
-2.0 (0.4453) |
0.2899 |
|
Échelle ONLS (p) |
0.0006 |
0.0876 |
0.4030 |
0.0033 |
0.0661 |
0.8884 |
|
Échelle Rankin (p) |
0.0006 |
0.0220 |
0.3542 |
0.0132 |
0.2543 |
0.8360 |
|
Échelle Rotterdam [delta (p)] |
+1.4 (0.0071) |
+1.3 (0.0342) |
0.6465 |
+1.1 (0.0342) |
+1.1 (0.0859) |
0.4056 |
|
SF-36 Qualité de vie |
+14.2 (0.0011) |
+16.7 (0.0008) |
0.3634 |
+11.1 (0.0091) |
+16.0 (0.1094) |
0.6518 |
*ITT : pendant l'étude (12 mois) ; PP : au cours de la phase de follow-up (6 mois)
Population pédiatrique
Les données publiées sur les études d'efficacité et de sécurité n'ont pas détecté de grandes différences entre les adultes et les enfants atteints de la même maladie.
5.2. Propriétés pharmacocinétiques
Elle se répartit de manière relativement rapide entre le plasma et le liquide extravasculaire ; après environ 3-5 jours, on atteint un équilibre entre les compartiments intra- et extravasculaire.
L'immunoglobuline humaine normale a une demi-vie d'environ 26 jours. Cette demi-vie peut varier d'un patient à l'autre, particulièrement dans les déficits immunitaires primitifs.
Les IgG et les complexes d'IgG sont catabolisés dans les cellules du système réticulo-endothélial.
Population pédiatrique
Les données publiées sur les études de pharmacocinétique n'ont pas mis en évidence de différences entre les adultes et les enfants atteints de la même maladie.
Aucune donnée n'est disponible quant aux propriétés pharmacocinétiques chez les patients pédiatriques souffrant de PIDC.
5.3. Données de sécurité préclinique
Les immunoglobulines sont des constituants normaux de l'organisme humain. En outre, étant donné que dans les études sur les animaux, l'administration d'immunoglobulines peut conduire à la formation d'anticorps, les données de sécurité précliniques sont limitées. Quoi qu'il en soit, les quelques études effectuées sur les animaux n'ont montré aucun risque particulier pour l'homme, sur la base d'études de toxicité aiguë et subaiguë.
Eau pour préparations injectables.
3 ans
Une fois que le récipient pour la perfusion a été ouvert, le contenu doit être immédiatement utilisé.
6.4. Précautions particulières de conservation
À conserver au réfrigérateur (entre 2 °C et 8 °C). Conserver le flacon dans l'emballage extérieur.
Avant leur utilisation et avant leur date de péremption, les flacons de 50, 100 et 200 mL peuvent être conservés à une température ambiante ne dépassant pas 25 °C, jusqu'à un maximum de 6 mois consécutifs. Passé cette période, le produit doit être éliminé. Dans tous les cas, le produit ne peut plus être remis au réfrigérateur sil a été conservé à température ambiante.
La date de début de la conservation à la température ambiante doit être inscrite sur la boîte externe.
Ne pas congeler.
6.5. Nature et contenu de l'emballage extérieur
20 mL de solution dans un flacon unique (verre de type I) avec bouchon (caoutchouc en halogénobutyle).
50 mL, 100 mL et 200 mL de solution dans un flacon unique (verre de type I), avec bouchon (caoutchouc en halogénobutyle), étiquette avec suspensoir extensible intégré (flacon + suspensoir extensible).
Emballages :
Emballages individuels :
1 flacon contenant 1 g/20 mL
1 flacon contenant 2,5 g/50 mL
1 flacon contenant 5 g/100 mL
1 flacon contenant 10 g/200 mL
Conditionnements multiples :
Conditionnement multiple de 2 boîtes de 1 flacon de 10 g/200 mL.
Conditionnement multiple de 3 boîtes de 1 flacon de 10 g/200 mL.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières délimination et de manipulation
Ce produit doit être amené à température ambiante ou à la température du corps avant utilisation.
La solution doit être limpide ou légèrement opalescente, incolore ou jaune pâle.
Ne pas utiliser de solutions troubles ou contenant des dépôts.
Avant l'administration, inspecter visuellement la solution à la recherche de corpuscules ou d'altérations de la couleur.
Flacons 50 mL, 100 mL et 200 mL
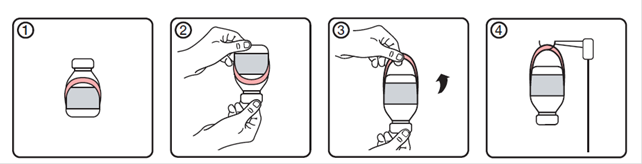
Mode demploi du suspensoir extensible
|
|
1. État initial du flacon avec l'étiquette porte-flacon
2. Retourner le flacon
3. Faites pivoter vers le haut le bord inférieur de l'étiquette porte-flacon pour létendre
4. Suspendre le flacon au support.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE LAUTORISATION DE MISE SUR LE MARCHE
Loc. Ai Conti
55051 Castelvecchio Pascoli, Barga (Lucca)
ITALIE
8. NUMERO(S) DAUTORISATION DE MISE SUR LE MARCHE
· 34009 302 667 9 3 : 20 mL en flacon (verre).
· 34009 302 668 0 9 : 50 mL en flacon (verre) avec suspensoir.
· 34009 302 668 1 6 : 100 mL en flacon (verre) avec suspensoir.
· 34009 302 668 2 3 : 200 mL en flacon (verre) avec suspensoir.
· 34009 302 668 3 0 : Conditionnement multiple de 2 (2 boîtes de 1) flacons (verre) de 200 mL avec suspensoir.
· 34009 302 668 5 4 : Conditionnement multiple de 3 (3 boîtes de 1) flacons (verre) de 200 mL avec suspensoir.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE LAUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière.
La prescription par un médecin exerçant dans un établissement de transfusion sanguine autorisé à dispenser des médicaments aux malades qui y sont traités est également autorisée.